

Wernig Laboratory

October 9, 2024: Welcome new postdoc Kirill to the lab!
Sep 17, 2024: Jinzhao's paper accepted in principle!
Jul 11, 2024: Gernot's paper published in Nature Communications!
March 21, 2024: Marius Mader's paper comes out in Nature Neuroscience!
Oct 10, 2023: Yongjin receives the Sammy Kuo Award from the Neuroscience Institute - CONGRATULATIONS!
Yongjin's paper on cell therapy in a mouse AD model is published in Cell Stem Cells
Our lab is generally interested in the molecular mechanisms that determine cell fates
Recently, we have identified a pool of transcription factors that are sufficient to convert skin fibroblasts directly into functional neuronal cells that we termed induced neuronal (iN) cells. This was a surprising finding and indicated that direct lineage reprogramming may be applicable to many somatic cell types and many different directions. Indeed, following our work others have identified transcription factors that could induce cardiomyocytes, blood progenitors, and hepatocytes from fibroblasts.
We are now focussing on two major aspects of iN and iPS cell reprogramming:
(i) we are fascinated by the puzzle how a hand full of transcription factors can so efficiently reprogram the entire epigenome of a cell so that it changes identity. To that end we are applying genome-wide expression analysis, chromatin immunoprecipitation, protein biochemistry, proteomics and functional screens.
(ii) it is equally exciting to now use reprogramming methods as tools to study or treat certain diseases. iPS cells have the great advantage that they can easily be genetically manipulated rendering them ideal for treating monogenetic disorders when combined with cell transplantation-based therapies. In particular we are working on Dystrophic Epidermolysis Bullosa in collaboration with Stanford's Dermatology Department. An exciting application of iN cell technology will be to try modeling neurological diseases in vitro. We perform both mouse and human experiments hoping to identify quantifiable phenotypes correlated with genotype and in a second step evaluate whether this assay could be used to discover novel drugs improve the disease progression.
Wernig Lab Research
Overview
Our lab is interested in the molecular mechanisms that define neural lineage identity focusing on transcription factors and chromatin biology. We use cellular reprogramming to understand how neurons are induced, how they mature and maintain their identity. Reprogramming also allows us to generate a novel tool box to study human neuronal and glial cell biology which become powerful human disease models in combination with genetic engineering. We further seek to develop reprogramming & genetic engineering approaches towards stem cell-based therapies. Finally, we study microglia-neuron interactions with the ultimate goal to understand the brain's immune system in health and disease and to exploit microglia for therapeutic and regenerative purposes.
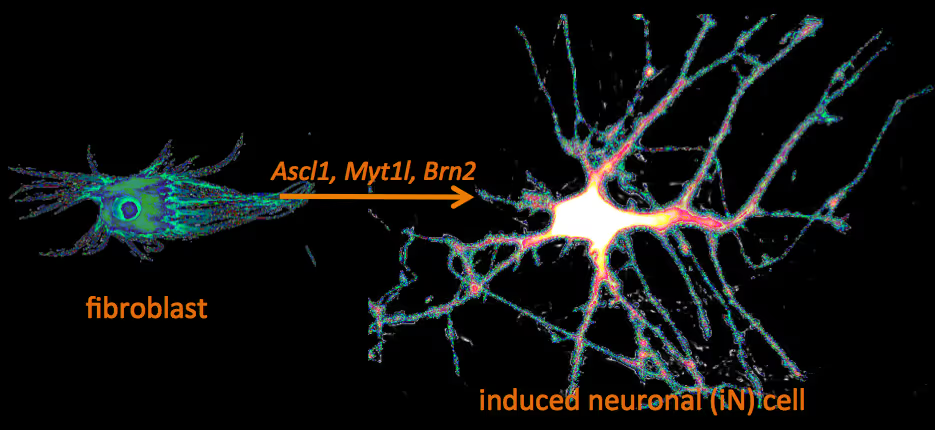
Human neuronal cell disease modeling
Neurosychiatric diseases like autism and schizophrenia are highly complex brain disorders difficult to model in mice in part due to complex genetic etiology and sometimes affecting human-specific genes. We develop novel human cell models to investigate disease-relevant cell biological phenomena.
Generation of defined human neuronal cell types to study neuronal cell biology
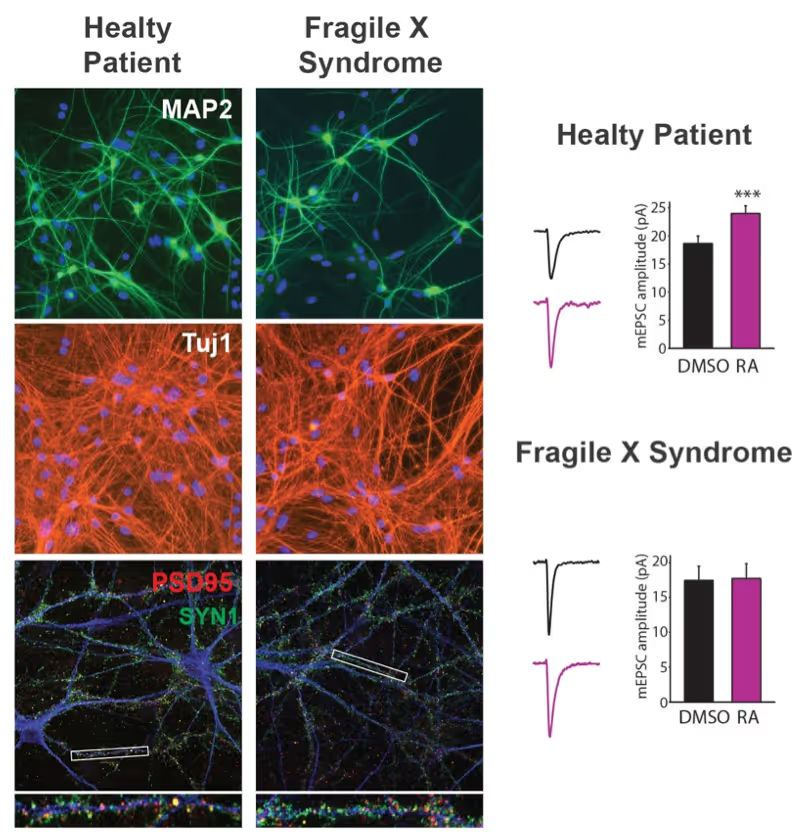
We have and continue to develop protocols to generate specific types of neurons such as pure glutamatergic and pure GABAergic neurons from human pluripotent stem cells using transcription factors. In combination with genetic engineering or deriving iPS cells from patients, we then interrogate the cell biology of human neurons that carry disease-causing mutations. A particular focus is on synaptic function as shown in the figure on the right on Fragile X Syndrome neurons in collaboration with Lu Chen and Tom Südhof's laboratories.
Making neurons from blood
The ability to generate functional induced neuronal cells from distantly related somatic cell types is fascinating but also offers the opportunity to obtain neurons from a larger cohort of human subjects. In particular blood is readily available and we showed can be efficiently converted into functional neurons from young and aged donors.
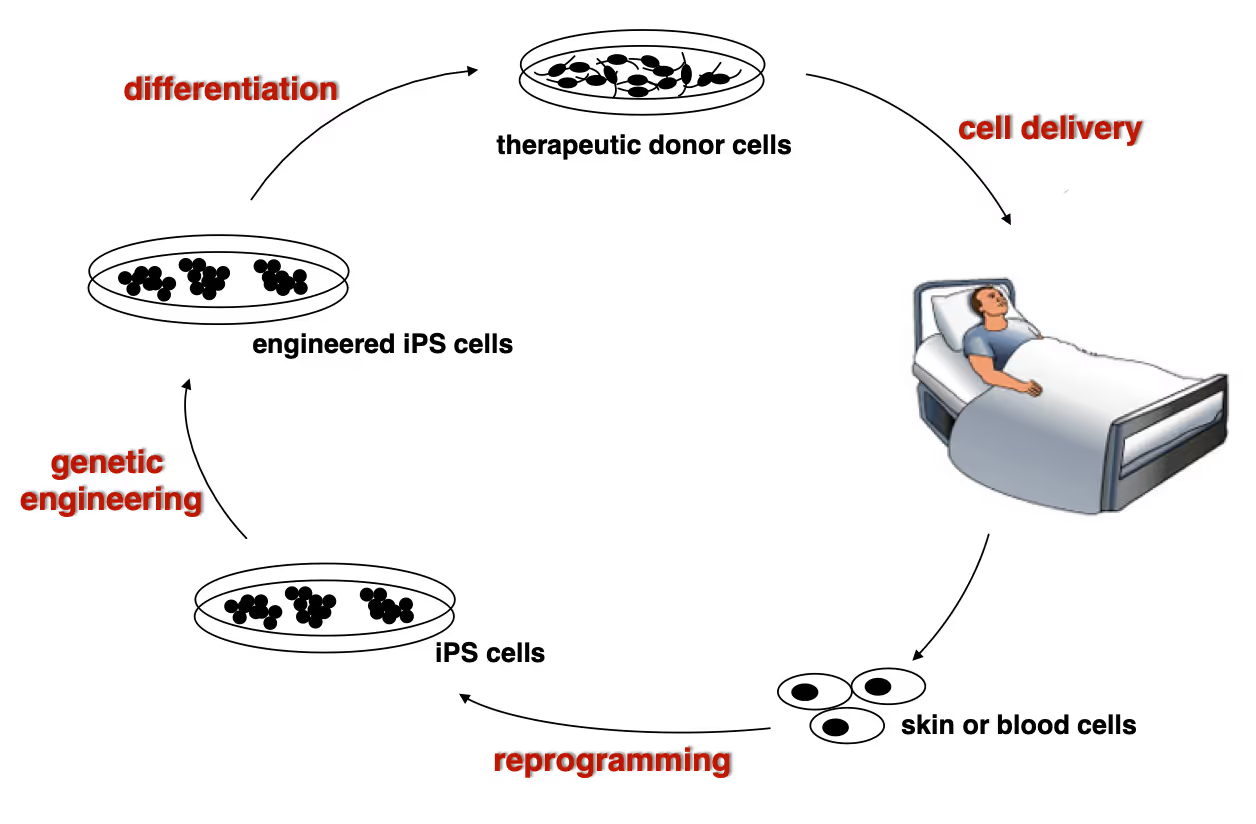
Developing next generation cell therapies
The combination of reprogramming and gene editing is truly powerful as it provides exciting new possibilities to generate cells that can be transplanted and have disease modifying activity. We currently apply this approach to restore mono-genetic diseases, but our vision goes beyond simple regenerative medicine. We will be able to genetically engineer designer cells that functionally integrate into diseased tissue equipped with sensing and intelligent disease-response mechanisms.
Towards a Phase 1 clinical trial for the fatal skin disease Epidermolysis Bullosa
Dystrophic Epidermolysis Bullosa is a severe, blistering monogenetic skin disease caused by mutations in the gene coding for type VII collagen. We have developed a 1-step gene editing/iPS cell reprogramming method to rapidly generate patient iPS cells corrected for their disease-causing mutations in the Collagen7a1 gene. In collaboration with dermatologist Tony Oro we are developing a cell manufacturing process compatible with Good Manufacturing Procedures (GMP) to obtain FDA-approval for a first in man Phase I clinical trial with with a genetically engineered iPS cell product.
Exploiting glia cell transplantation to treat neurodegenerative disease
Both oligodendrocyte precursor cells as well as microglia can efficiently repopulate the brain. We are interested in exploiting the properties of these cells to develop novel cell therapies for the brain either to use the transplanted cells to restore function such as myelination, to alter the function of transplanted cells for therepeutic benefit, to use the cells as vehicles for therapeutic molecules, or ultimately to develop designer cells that are engineered with genetic synthetic biology circuits to sense and interfere with disease processes of the brain.
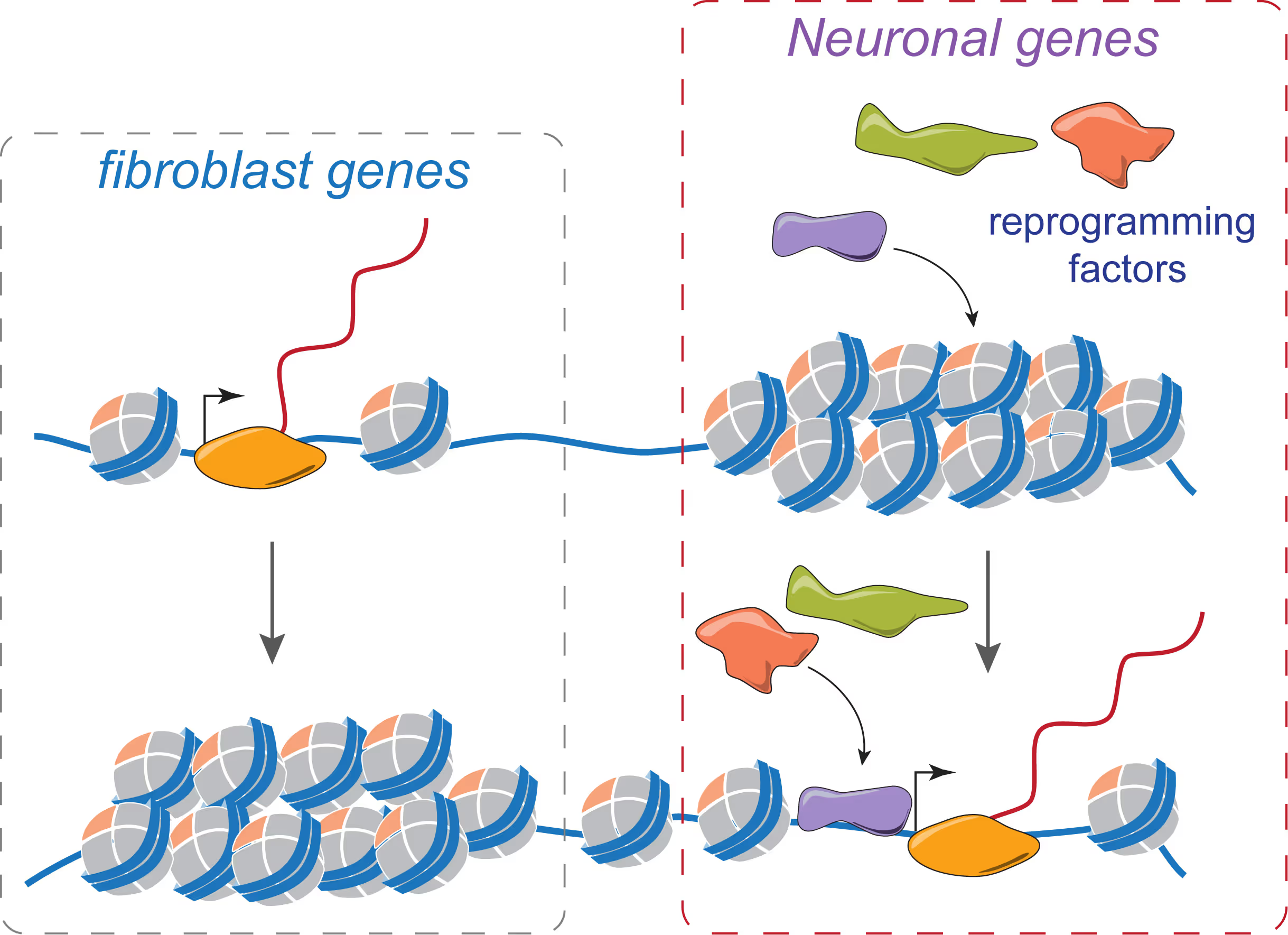
Mechanisms of neural cell lineage identity
We are interested in the molecular mechanisms that define neuronal and glial cell identity. We found sets of transcription factors that can convert fibroblasts or lymphocytes into neurons and oligodendrocytes. These factors are also operational during normal development and are largely responsible to induce terminal lineages from progenitor cells.
"On target" pioneer factors and chromatin remodeling during neuronal induction
We found that Ascl1, one of our reprogramming factors, has a unique ability to access its physiological targets even in fibroblasts where these sites are in a closed chromatin configuration. We are fascinated by this "on target" pioneering property and are investigating how Ascl1 can access its target sites in an unfavorable chromatin environment and how it then remodels the chromatin at these sites to activate the neuronal transcriptional program.
Maintenance of neuronal identity
Once neurons are made, there ought to be also mechanisms that maintain neuronal identity. We stumbled upon a novel repressive mechanism: The neuronal-specific transcription factor Myt1l continuosly represses many non-neuronal programs in neurons leaving the neuronal program open to activate by other factors and thereby ensuring stable neuronal gene expression. Myt1l was also recently found to be mutated in autism and schizophrenia.
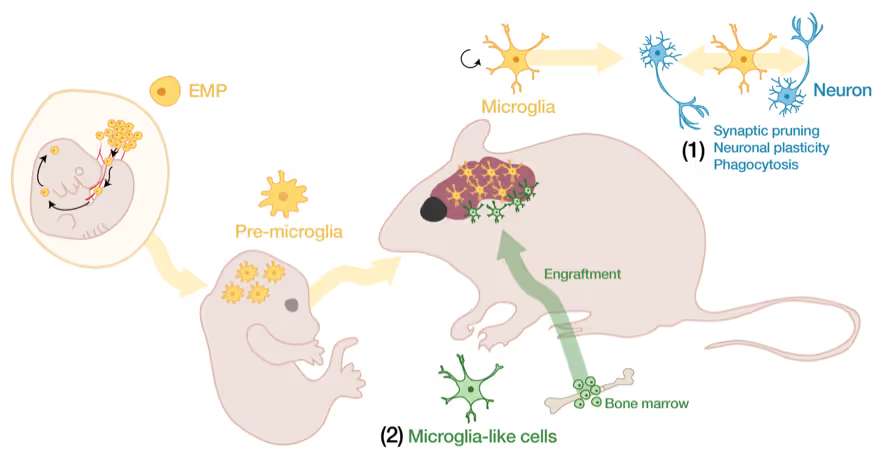
Microglia-neuron interactions in the healthy and diseased brain
Microglia, the brain's resident immune cells, are fascinating cells. They are derived from yolk sac progenitor cells early during development, are long-lived, and are not exchanged from bone marrow progenitor cells under physiological conditions. Microglia have been implicated in synaptic pruning, adult neurogenesis, and various brain diseases including Alzheimer's disease and Schizophrenia.
Developing an efficient microglia replacement system
We have developed a method to efficiently replace endogenous microglia from circulating cells without genetic manipulation. This does not happen physiologically but under certain conditions peripheral blood cells cross the blood-brain-barrier, migrate into the brain parenchyma and replace endogenous cells. We are investigating the cellular and molecular signals that enable circulating cells to invade the brain in order to further improve microglia replacement strategies.
The role of microglia in the normal and the diseased brain
Our ability to replace microglia provides us with a powerful tool to functionally perturb microglia function in normal and disease states. E.g. the microglial gene TREM2 is a strong Alzheimer's disease risk gene, but major questions about the neuro-immune interplay in the context of neurodegeneration and aging remain unsolved. Microglia replacement also provides an exciting prospect to develop novel cell therapies for a variety of brain diseases including enzyme deficiency syndromes, neurodegeneration, and brain tumors.
Lab Gene Expression Data
Publications
Margaret G Guo, David L Reynolds, Cheen E Ang, Yingfei Liu, Yang Zhao, Laura K H Donohue, Zurab Siprashvili, Xue Yang, Yongjin Yoo, Smarajit Mondal, Audrey Hong, Jessica Kain, Lindsey Meservey, Tania Fabo, Ibtihal Elfaki, Laura N Kellman, Nathan S Abell, Yash Pershad, Vafa Bayat, Payam Etminani, Mark Holodniy, Daniel H Geschwind, Stephen B Montgomery, Laramie E Duncan, Alexander E Urban, Russ B Altman, Marius Wernig, Paul A Khavari
Noncoding variants of presumed regulatory function contribute to the heritability of neuropsychiatric disease. A total of 2,221 noncoding variants connected to risk for ten neuropsychiatric disorders, including autism spectrum disorder, attention deficit hyperactivity disorder, bipolar disorder, borderline personality disorder, major depression, generalized anxiety disorder, panic disorder, post-traumatic stress disorder, obsessive-compulsive disorder and schizophrenia, were studied in developing human neural cells. Integrating epigenomic and transcriptomic data with massively parallel reporter assays identified differentially-active single-nucleotide variants (daSNVs) in specific neural cell types. Expression-gene mapping, network analyses and chromatin looping nominated candidate disease-relevant target genes modulated by these daSNVs. Follow-up integration of daSNV gene editing with clinical cohort analyses suggested that magnesium transport dysfunction may increase neuropsychiatric disease risk and indicated that common genetic pathomechanisms may mediate specific symptoms that are shared across multiple neuropsychiatric diseases.
Noncoding variants of presumed regulatory function contribute to the heritability of neuropsychiatric disease. A total of 2,221 noncoding variants connected to risk for ten neuropsychiatric disorders, including autism spectrum disorder, attention deficit hyperactivity disorder, bipolar disorder, borderline personality disorder, major depression, generalized anxiety disorder, panic disorder, post-traumatic stress disorder, obsessive-compulsive disorder and schizophrenia, were studied in developing human neural cells. Integrating epigenomic and transcriptomic data with massively parallel reporter assays identified differentially-active single-nucleotide variants (daSNVs) in specific neural cell types. Expression-gene mapping, network analyses and chromatin looping nominated candidate disease-relevant target genes modulated by these daSNVs. Follow-up integration of daSNV gene editing with clinical cohort analyses suggested that magnesium transport dysfunction may increase neuropsychiatric disease risk and indicated that common genetic pathomechanisms may mediate specific symptoms that are shared across multiple neuropsychiatric diseases.
Yongjin Yoo, Gernot Neumayer, Yohei Shibuya, Marius Marc-Daniel Mader, Marius Wernig
Gintautas Vainorius, Maria Novatchkova, Georg Michlits, Juliane Christina Baar, Cecilia Raupach, Joonsun Lee, Ramesh Yelagandula, Marius Wernig, Ulrich Elling
Ascl1 and Ngn2, closely related proneural transcription factors, are able to convert mouse embryonic stem cells into induced neurons. Despite their similarities, these factors elicit only partially overlapping transcriptional programs, and it remains unknown whether cells are converted via distinct mechanisms. Here we show that Ascl1 and Ngn2 induce mutually exclusive side populations by binding and activating distinct lineage drivers. Furthermore, Ascl1 rapidly dismantles the pluripotency network and installs neuronal and trophoblast cell fates, while Ngn2 generates a neural stem cell-like intermediate supported by incomplete shutdown of the pluripotency network. Using CRISPR-Cas9 knockout screening, we find that Ascl1 relies more on factors regulating pluripotency and the cell cycle, such as Tcf7l1. In the absence of Tcf7l1, Ascl1 still represses core pluripotency genes but fails to exit the cell cycle. However, overexpression of Cdkn1c induces cell cycle exit and restores the generation of neurons. These findings highlight that cell type conversion can occur through two distinct mechanistic paths, even when induced by closely related transcription factors.
Ascl1 and Ngn2, closely related proneural transcription factors, are able to convert mouse embryonic stem cells into induced neurons. Despite their similarities, these factors elicit only partially overlapping transcriptional programs, and it remains unknown whether cells are converted via distinct mechanisms. Here we show that Ascl1 and Ngn2 induce mutually exclusive side populations by binding and activating distinct lineage drivers. Furthermore, Ascl1 rapidly dismantles the pluripotency network and installs neuronal and trophoblast cell fates, while Ngn2 generates a neural stem cell-like intermediate supported by incomplete shutdown of the pluripotency network. Using CRISPR-Cas9 knockout screening, we find that Ascl1 relies more on factors regulating pluripotency and the cell cycle, such as Tcf7l1. In the absence of Tcf7l1, Ascl1 still represses core pluripotency genes but fails to exit the cell cycle. However, overexpression of Cdkn1c induces cell cycle exit and restores the generation of neurons. These findings highlight that cell type conversion can occur through two distinct mechanistic paths, even when induced by closely related transcription factors.
Yingfei Liu, Jinzhao Wang, Thomas C Südhof, Marius Wernig
The production of induced neuronal (iN) cells from human embryonic stem cells (ESCs) and induced pluripotent stem cells by the forced expression of proneural transcription factors is rapid, efficient and reproducible. The ability to generate large numbers of human neurons in such a robust manner enables large-scale studies of human neural differentiation and neuropsychiatric diseases. Surprisingly, similar transcription factor-based approaches for converting mouse ESCs into iN cells have been challenging, primarily because of low cell survival. Here, we provide a detailed approach for the efficient and reproducible generation of functional iN cells from mouse ESC cultures by the genetically induced expression of neurogenin-2. The resulting iN cells display mature pre- and postsynaptic specializations and form synaptic networks. Our method provides the basis for studying neuronal development and enables the direct comparison of cellular phenotypes in mouse and human neurons generated in an equivalent way. The procedure requires 14 d and can be carried out by users with expertise in stem cell culture.
The production of induced neuronal (iN) cells from human embryonic stem cells (ESCs) and induced pluripotent stem cells by the forced expression of proneural transcription factors is rapid, efficient and reproducible. The ability to generate large numbers of human neurons in such a robust manner enables large-scale studies of human neural differentiation and neuropsychiatric diseases. Surprisingly, similar transcription factor-based approaches for converting mouse ESCs into iN cells have been challenging, primarily because of low cell survival. Here, we provide a detailed approach for the efficient and reproducible generation of functional iN cells from mouse ESC cultures by the genetically induced expression of neurogenin-2. The resulting iN cells display mature pre- and postsynaptic specializations and form synaptic networks. Our method provides the basis for studying neuronal development and enables the direct comparison of cellular phenotypes in mouse and human neurons generated in an equivalent way. The procedure requires 14 d and can be carried out by users with expertise in stem cell culture.
Yongjin Yoo, Gernot Neumayer, Yohei Shibuya, Marius Marc-Daniel Mader, Marius Wernig
Alzheimer's disease (AD) remains one of the grand challenges facing human society. Much controversy exists around the complex and multifaceted pathogenesis of this prevalent disease. Given strong human genetic evidence, there is little doubt, however, that microglia play an important role in preventing degeneration of neurons. For example, loss of function of the microglial gene Trem2 renders microglia dysfunctional and causes an early-onset neurodegenerative syndrome, and Trem2 variants are among the strongest genetic risk factors for AD. Thus, restoring microglial function represents a rational therapeutic approach. Here, we show that systemic hematopoietic cell transplantation followed by enhancement of microglia replacement restores microglial function in a Trem2 mutant mouse model of AD.
Alzheimer's disease (AD) remains one of the grand challenges facing human society. Much controversy exists around the complex and multifaceted pathogenesis of this prevalent disease. Given strong human genetic evidence, there is little doubt, however, that microglia play an important role in preventing degeneration of neurons. For example, loss of function of the microglial gene Trem2 renders microglia dysfunctional and causes an early-onset neurodegenerative syndrome, and Trem2 variants are among the strongest genetic risk factors for AD. Thus, restoring microglial function represents a rational therapeutic approach. Here, we show that systemic hematopoietic cell transplantation followed by enhancement of microglia replacement restores microglial function in a Trem2 mutant mouse model of AD.
Ching-Chieh Chou, Ryan Vest, Miguel A Prado, Joshua Wilson-Grady, Joao A Paulo, Yohei Shibuya, Patricia Moran-Losada, Ting-Ting Lee, Jian Luo, Steven P Gygi, Jeffery W Kelly, Daniel Finley, Marius Wernig, Tony Wyss-Coray, Judith Frydman
The role of proteostasis and organelle homeostasis dysfunction in human aging and Alzheimer's disease (AD) remains unclear. Analyzing proteome-wide changes in human donor fibroblasts and their corresponding transdifferentiated neurons (tNeurons), we find aging and AD synergistically impair multiple proteostasis pathways, most notably lysosomal quality control (LQC). In particular, we show that ESCRT-mediated lysosomal repair defects are associated with both sporadic and PSEN1 familial AD. Aging- and AD-linked defects are detected in fibroblasts but highly exacerbated in tNeurons, leading to enhanced neuronal vulnerability, unrepaired lysosomal damage, inflammatory factor secretion and cytotoxicity. Surprisingly, tNeurons from aged and AD donors spontaneously develop amyloid-β inclusions co-localizing with LQC markers, LAMP1/2-positive lysosomes and proteostasis factors; we observe similar inclusions in brain tissue from AD patients and APP-transgenic mice. Importantly, compounds enhancing lysosomal function broadly ameliorate these AD-associated pathologies. Our findings establish cell-autonomous LQC dysfunction in neurons as a central vulnerability in aging and AD pathogenesis.
The role of proteostasis and organelle homeostasis dysfunction in human aging and Alzheimer's disease (AD) remains unclear. Analyzing proteome-wide changes in human donor fibroblasts and their corresponding transdifferentiated neurons (tNeurons), we find aging and AD synergistically impair multiple proteostasis pathways, most notably lysosomal quality control (LQC). In particular, we show that ESCRT-mediated lysosomal repair defects are associated with both sporadic and PSEN1 familial AD. Aging- and AD-linked defects are detected in fibroblasts but highly exacerbated in tNeurons, leading to enhanced neuronal vulnerability, unrepaired lysosomal damage, inflammatory factor secretion and cytotoxicity. Surprisingly, tNeurons from aged and AD donors spontaneously develop amyloid-β inclusions co-localizing with LQC markers, LAMP1/2-positive lysosomes and proteostasis factors; we observe similar inclusions in brain tissue from AD patients and APP-transgenic mice. Importantly, compounds enhancing lysosomal function broadly ameliorate these AD-associated pathologies. Our findings establish cell-autonomous LQC dysfunction in neurons as a central vulnerability in aging and AD pathogenesis.
Marius Marc-Daniel Mader, Alan Napole, Danwei Wu, Yohei Shibuya, Alexa Scavetti, Aulden Foltz, Micaiah Atkins, Oliver Hahn, Yongjin Yoo, Ron Danziger, Christina Tan, Tony Wyss-Coray, Lawrence Steinman, Marius Wernig
Multiple sclerosis (MS) is an autoimmune disease associated with inflammatory demyelination in the central nervous system (CNS). Autologous hematopoietic cell transplantation (HCT) is under investigation as a promising therapy for treatment-refractory MS. Here we identify a reactive myeloid state in chronic experimental autoimmune encephalitis (EAE) mice and MS patients that is surprisingly associated with neuroprotection and immune suppression. HCT in EAE mice leads to an enhancement of this myeloid state, as well as clinical improvement, reduction of demyelinated lesions, suppression of cytotoxic T cells, and amelioration of reactive astrogliosis reflected in reduced expression of EAE- associated gene signatures in oligodendrocytes and astrocytes. Further enhancement of myeloid cell incorporation into the CNS following a modified HCT protocol results in an even more consistent therapeutic effect corroborated by additional amplification of HCT-induced transcriptional changes, underlining myeloid-derived beneficial effects in the chronic phase of EAE. Replacement or manipulation of CNS myeloid cells thus represents an intriguing therapeutic direction for inflammatory demyelinating disease.
Multiple sclerosis (MS) is an autoimmune disease associated with inflammatory demyelination in the central nervous system (CNS). Autologous hematopoietic cell transplantation (HCT) is under investigation as a promising therapy for treatment-refractory MS. Here we identify a reactive myeloid state in chronic experimental autoimmune encephalitis (EAE) mice and MS patients that is surprisingly associated with neuroprotection and immune suppression. HCT in EAE mice leads to an enhancement of this myeloid state, as well as clinical improvement, reduction of demyelinated lesions, suppression of cytotoxic T cells, and amelioration of reactive astrogliosis reflected in reduced expression of EAE- associated gene signatures in oligodendrocytes and astrocytes. Further enhancement of myeloid cell incorporation into the CNS following a modified HCT protocol results in an even more consistent therapeutic effect corroborated by additional amplification of HCT-induced transcriptional changes, underlining myeloid-derived beneficial effects in the chronic phase of EAE. Replacement or manipulation of CNS myeloid cells thus represents an intriguing therapeutic direction for inflammatory demyelinating disease.
Gernot Neumayer, Jessica L Torkelson, Shengdi Li, Kelly McCarthy, Hanson H Zhen, Madhuri Vangipuram, Joanna Jackow, Avina Rami, Corey Hansen, Zongyou Guo, Sadhana Gaddam, Alberto Pappalardo, Lingjie Li, Amber Cramer, Kevin R Roy, Thuylinh Michelle Nguyen, Koji Tanabe, Patrick S McGrath, Anna Bruckner, Ganna Bilousova, Dennis Roop, Irene Bailey, Jean Y Tang, Angela Christiano, Lars M Steinmetz, Marius Wernig, Anthony E Oro
BACKGROUND: Gene editing in induced pluripotent stem (iPS) cells has been hailed to enable new cell therapies for various monogenetic diseases including dystrophic epidermolysis bullosa (DEB). However, manufacturing, efficacy and safety roadblocks have limited the development of genetically corrected, autologous iPS cell-based therapies. METHODS: We developed Dystrophic Epidermolysis Bullosa Cell Therapy (DEBCT), a new generation GMP-compatible (cGMP), reproducible, and scalable platform to produce autologous clinical-grade iPS cell-derived organotypic induced skin composite (iSC) grafts to treat incurable wounds of patients lacking type VII collagen (C7). DEBCT uses a combined high-efficiency reprogramming and CRISPR-based genetic correction single step to generate genome scar- free, COL7A1 corrected clonal iPS cells from primary patient fibroblasts. Validated iPS cells are converted into epidermal, dermal and melanocyte progenitors with a novel 2D organoid differentiation protocol, followed by CD49f enrichment and expansion to minimize maturation heterogeneity. iSC product characterization by single cell transcriptomics was followed by mouse xenografting for disease correcting activity at 1 month and toxicology analysis at 1-6 months. Culture-acquired mutations, potential CRISPR-off targets, and cancer-driver variants were evaluated by targeted and whole genome sequencing. FINDINGS: iPS cell-derived iSC grafts were reproducibly generated from four recessive DEB patients with different pathogenic mutations. Organotypic iSC grafts onto immune-compromised mice developed into stable stratified skin with functional C7 restoration. Single cell transcriptomic characterization of iSCs revealed prominent holoclone stem cell signatures in keratinocytes and the recently described Gibbin-dependent signature in dermal fibroblasts. The latter correlated with enhanced graftability. Multiple orthogonal sequencing and subsequent computational approaches identified random and non-oncogenic mutations introduced by the manufacturing process. Toxicology revealed no detectable tumors after 3-6 months in DEBCT- treated mice. INTERPRETATION: DEBCT successfully overcomes previous roadblocks and represents a robust, scalable, and safe cGMP manufacturing platform for production of a CRISPR-corrected autologous organotypic skin graft to heal DEB patient wounds.
BACKGROUND: Gene editing in induced pluripotent stem (iPS) cells has been hailed to enable new cell therapies for various monogenetic diseases including dystrophic epidermolysis bullosa (DEB). However, manufacturing, efficacy and safety roadblocks have limited the development of genetically corrected, autologous iPS cell-based therapies. METHODS: We developed Dystrophic Epidermolysis Bullosa Cell Therapy (DEBCT), a new generation GMP-compatible (cGMP), reproducible, and scalable platform to produce autologous clinical-grade iPS cell-derived organotypic induced skin composite (iSC) grafts to treat incurable wounds of patients lacking type VII collagen (C7). DEBCT uses a combined high-efficiency reprogramming and CRISPR-based genetic correction single step to generate genome scar- free, COL7A1 corrected clonal iPS cells from primary patient fibroblasts. Validated iPS cells are converted into epidermal, dermal and melanocyte progenitors with a novel 2D organoid differentiation protocol, followed by CD49f enrichment and expansion to minimize maturation heterogeneity. iSC product characterization by single cell transcriptomics was followed by mouse xenografting for disease correcting activity at 1 month and toxicology analysis at 1-6 months. Culture-acquired mutations, potential CRISPR-off targets, and cancer-driver variants were evaluated by targeted and whole genome sequencing. FINDINGS: iPS cell-derived iSC grafts were reproducibly generated from four recessive DEB patients with different pathogenic mutations. Organotypic iSC grafts onto immune-compromised mice developed into stable stratified skin with functional C7 restoration. Single cell transcriptomic characterization of iSCs revealed prominent holoclone stem cell signatures in keratinocytes and the recently described Gibbin-dependent signature in dermal fibroblasts. The latter correlated with enhanced graftability. Multiple orthogonal sequencing and subsequent computational approaches identified random and non-oncogenic mutations introduced by the manufacturing process. Toxicology revealed no detectable tumors after 3-6 months in DEBCT- treated mice. INTERPRETATION: DEBCT successfully overcomes previous roadblocks and represents a robust, scalable, and safe cGMP manufacturing platform for production of a CRISPR-corrected autologous organotypic skin graft to heal DEB patient wounds.
Bahareh Haddad Derafshi, Tamas Danko, Soham Chanda, Pedro J Batista, Ulrike Litzenburger, Qian Yi Lee, Yi Han Ng, Anu Sebin, Howard Y Chang, Thomas C Südhof, Marius Wernig
The chromodomain helicase DNA-binding protein CHD8 is the most frequently mutated gene in autism spectrum disorder. Despite its prominent disease involvement, little is known about its molecular function in the human brain. CHD8 is a chromatin regulator which binds to the promoters of actively transcribed genes through genomic targeting mechanisms which have yet to be fully defined. By generating a conditional loss-of-function and an endogenously tagged allele in human pluripotent stem cells, we investigated the molecular function and the interaction of CHD8 with chromatin in human neurons. Chromatin accessibility analysis and transcriptional profiling revealed that CHD8 functions as a transcriptional activator at its target genes in human neurons. Furthermore, we found that CHD8 chromatin targeting is cell context-dependent. In human neurons, CHD8 preferentially binds at ETS motif-enriched promoters. This enrichment is particularly prominent on the promoters of genes whose expression significantly changes upon the loss of CHD8. Indeed, among the ETS transcription factors, we identified ELK1 as being most highly correlated with CHD8 expression in primary human fetal and adult cortical neurons and most highly expressed in our stem cell-derived neurons. Remarkably, ELK1 was necessary to recruit CHD8 specifically to ETS motif-containing sites. These findings imply that ELK1 and CHD8 functionally cooperate to regulate gene expression and chromatin states at MAPK/ERK target genes in human neurons. Our results suggest that the MAPK/ERK/ELK1 axis potentially contributes to the pathogenesis caused by CHD8 mutations in human neurodevelopmental disorders.
The chromodomain helicase DNA-binding protein CHD8 is the most frequently mutated gene in autism spectrum disorder. Despite its prominent disease involvement, little is known about its molecular function in the human brain. CHD8 is a chromatin regulator which binds to the promoters of actively transcribed genes through genomic targeting mechanisms which have yet to be fully defined. By generating a conditional loss-of-function and an endogenously tagged allele in human pluripotent stem cells, we investigated the molecular function and the interaction of CHD8 with chromatin in human neurons. Chromatin accessibility analysis and transcriptional profiling revealed that CHD8 functions as a transcriptional activator at its target genes in human neurons. Furthermore, we found that CHD8 chromatin targeting is cell context-dependent. In human neurons, CHD8 preferentially binds at ETS motif-enriched promoters. This enrichment is particularly prominent on the promoters of genes whose expression significantly changes upon the loss of CHD8. Indeed, among the ETS transcription factors, we identified ELK1 as being most highly correlated with CHD8 expression in primary human fetal and adult cortical neurons and most highly expressed in our stem cell-derived neurons. Remarkably, ELK1 was necessary to recruit CHD8 specifically to ETS motif-containing sites. These findings imply that ELK1 and CHD8 functionally cooperate to regulate gene expression and chromatin states at MAPK/ERK target genes in human neurons. Our results suggest that the MAPK/ERK/ELK1 axis potentially contributes to the pathogenesis caused by CHD8 mutations in human neurodevelopmental disorders.
Justyna A Janas, Lichao Zhang, Jacklyn H Luu, Janos Demeter, Lingjun Meng, Samuele G Marro, Moritz Mall, Nancie A Mooney, Katie Schaukowitch, Yi Han Ng, Nan Yang, Yuhao Huang, Gernot Neumayer, Or Gozani, Joshua E Elias, Peter K Jackson, Marius Wernig
Cell lineage specification is accomplished by a concerted action of chromatin remodeling and tissue-specific transcription factors. However, the mechanisms that induce and maintain appropriate lineage-specific gene expression remain elusive. Here, we used an unbiased proteomics approach to characterize chromatin regulators that mediate the induction of neuronal cell fate. We found that Tip60 acetyltransferase is essential to establish neuronal cell identity partly via acetylation of the histone variant H2A.Z. Despite its tight correlation with gene expression and active chromatin, loss of H2A.Z acetylation had little effect on chromatin accessibility or transcription. Instead, loss of Tip60 and acetyl-H2A.Z interfered with H3K4me3 deposition and activation of a unique subset of silent, lineage-restricted genes characterized by a bivalent chromatin configuration at their promoters. Altogether, our results illuminate the mechanisms underlying bivalent chromatin activation and reveal that H2A.Z acetylation regulates neuronal fate specification by establishing epigenetic competence for bivalent gene activation and cell lineage transition.
Cell lineage specification is accomplished by a concerted action of chromatin remodeling and tissue-specific transcription factors. However, the mechanisms that induce and maintain appropriate lineage-specific gene expression remain elusive. Here, we used an unbiased proteomics approach to characterize chromatin regulators that mediate the induction of neuronal cell fate. We found that Tip60 acetyltransferase is essential to establish neuronal cell identity partly via acetylation of the histone variant H2A.Z. Despite its tight correlation with gene expression and active chromatin, loss of H2A.Z acetylation had little effect on chromatin accessibility or transcription. Instead, loss of Tip60 and acetyl-H2A.Z interfered with H3K4me3 deposition and activation of a unique subset of silent, lineage-restricted genes characterized by a bivalent chromatin configuration at their promoters. Altogether, our results illuminate the mechanisms underlying bivalent chromatin activation and reveal that H2A.Z acetylation regulates neuronal fate specification by establishing epigenetic competence for bivalent gene activation and cell lineage transition.
Ziming Luo, Kun-Che Chang, Suqian Wu, Catalina Sun, Xin Xia, Michael Nahmou, Minjuan Bian, Rain R Wen, Ying Zhu, Sahil Shah, Bogdan Tanasa, Marius Wernig, Jeffrey L Goldberg
Retinal ganglion cell (RGC) replacement therapy could restore vision in glaucoma and other optic neuropathies. We developed a rapid protocol for directly induced RGC (iRGC) differentiation from human stem cells, leveraging overexpression of NGN2. Neuronal morphology and neurite growth were observed within 1 week of induction; characteristic RGC-specific gene expression confirmed identity. Calcium imaging demonstrated γ-aminobutyric acid (GABA)-induced excitation characteristic of immature RGCs. Single-cell RNA sequencing showed more similarities between iRGCs and early-stage fetal human RGCs than retinal organoid-derived RGCs. Intravitreally transplanted iRGCs survived and migrated into host retinas independent of prior optic nerve trauma, but iRGCs protected host RGCs from neurodegeneration. These data demonstrate rapid iRGC generation in vitro into an immature cell with high similarity to human fetal RGCs and capacity for retinal integration after transplantation and neuroprotective function after optic nerve injury. The simplicity of this system may benefit translational studies on human RGCs.
Retinal ganglion cell (RGC) replacement therapy could restore vision in glaucoma and other optic neuropathies. We developed a rapid protocol for directly induced RGC (iRGC) differentiation from human stem cells, leveraging overexpression of NGN2. Neuronal morphology and neurite growth were observed within 1 week of induction; characteristic RGC-specific gene expression confirmed identity. Calcium imaging demonstrated γ-aminobutyric acid (GABA)-induced excitation characteristic of immature RGCs. Single-cell RNA sequencing showed more similarities between iRGCs and early-stage fetal human RGCs than retinal organoid-derived RGCs. Intravitreally transplanted iRGCs survived and migrated into host retinas independent of prior optic nerve trauma, but iRGCs protected host RGCs from neurodegeneration. These data demonstrate rapid iRGC generation in vitro into an immature cell with high similarity to human fetal RGCs and capacity for retinal integration after transplantation and neuroprotective function after optic nerve injury. The simplicity of this system may benefit translational studies on human RGCs.
Bo Zhou, Jacqueline G Lu, Alberto Siddu, Marius Wernig, Thomas C Südhof
Mutations in β-amyloid (Aβ) precursor protein (APP ) cause familial Alzheimer's disease (AD) probably by enhancing Aβ peptides production from APP. An antibody targeting Aβ (aducanumab) was approved as an AD treatment; however, some Aβ antibodies have been reported to accelerate, instead of ameliorating, cognitive decline in individuals with AD. Using conditional APP mutations in human neurons for perfect isogenic controls and translational relevance, we found that the APP -Swedish mutation in familial AD increased synapse numbers and synaptic transmission, whereas the APP deletion decreased synapse numbers and synaptic transmission. Inhibition of BACE1, the protease that initiates Aβ production from APP, lowered synapse numbers, suppressed synaptic transmission in wild-type neurons, and occluded the phenotype of APP -Swedish-mutant neurons. Modest elevations of Aβ, conversely, elevated synapse numbers and synaptic transmission. Thus, the familial AD-linked APP -Swedish mutation under physiologically relevant conditions increased synaptic connectivity in human neurons via a modestly enhanced production of Aβ. These data are consistent with the relative inefficacy of BACE1 and anti-Aβ treatments in AD and the chronic nature of AD pathogenesis, suggesting that AD pathogenesis is not simply caused by overproduction of toxic Aβ but rather by a long-term effect of elevated Aβ concentrations.
Mutations in β-amyloid (Aβ) precursor protein (APP ) cause familial Alzheimer's disease (AD) probably by enhancing Aβ peptides production from APP. An antibody targeting Aβ (aducanumab) was approved as an AD treatment; however, some Aβ antibodies have been reported to accelerate, instead of ameliorating, cognitive decline in individuals with AD. Using conditional APP mutations in human neurons for perfect isogenic controls and translational relevance, we found that the APP -Swedish mutation in familial AD increased synapse numbers and synaptic transmission, whereas the APP deletion decreased synapse numbers and synaptic transmission. Inhibition of BACE1, the protease that initiates Aβ production from APP, lowered synapse numbers, suppressed synaptic transmission in wild-type neurons, and occluded the phenotype of APP -Swedish-mutant neurons. Modest elevations of Aβ, conversely, elevated synapse numbers and synaptic transmission. Thus, the familial AD-linked APP -Swedish mutation under physiologically relevant conditions increased synaptic connectivity in human neurons via a modestly enhanced production of Aβ. These data are consistent with the relative inefficacy of BACE1 and anti-Aβ treatments in AD and the chronic nature of AD pathogenesis, suggesting that AD pathogenesis is not simply caused by overproduction of toxic Aβ but rather by a long-term effect of elevated Aβ concentrations.
Kelsie Eichel, Takeshi Uenaka, Vivek Belapurkar, Rui Lu, Shouqiang Cheng, Joseph S Pak, Caitlin A Taylor, Thomas C Südhof, Robert Malenka, Marius Wernig, Engin Özkan, David Perrais, Kang Shen
Neurons are highly polarized cells that face the fundamental challenge of compartmentalizing a vast and diverse repertoire of proteins in order to function properly1 . The axon initial segment (AIS) is a specialized domain that separates a neuron's morphologically, biochemically and functionally distinct axon and dendrite compartments2,3 . How the AIS maintains polarity between these compartments is not fully understood. Here we find that in Caenorhabditis elegans, mouse, rat and human neurons, dendritically and axonally polarized transmembrane proteins are recognized by endocytic machinery in the AIS, robustly endocytosed and targeted to late endosomes for degradation. Forcing receptor interaction with the AIS master organizer, ankyrinG, antagonizes receptor endocytosis in the AIS, causes receptor accumulation in the AIS, and leads to polarity deficits with subsequent morphological and behavioural defects. Therefore, endocytic removal of polarized receptors that diffuse into the AIS serves as a membrane-clearance mechanism that is likely to work in conjunction with the known AIS diffusion-barrier mechanism to maintain neuronal polarity on the plasma membrane. Our results reveal a conserved endocytic clearance mechanism in the AIS to maintain neuronal polarity by reinforcing axonal and dendritic compartment membrane boundaries.
Neurons are highly polarized cells that face the fundamental challenge of compartmentalizing a vast and diverse repertoire of proteins in order to function properly1 . The axon initial segment (AIS) is a specialized domain that separates a neuron's morphologically, biochemically and functionally distinct axon and dendrite compartments2,3 . How the AIS maintains polarity between these compartments is not fully understood. Here we find that in Caenorhabditis elegans, mouse, rat and human neurons, dendritically and axonally polarized transmembrane proteins are recognized by endocytic machinery in the AIS, robustly endocytosed and targeted to late endosomes for degradation. Forcing receptor interaction with the AIS master organizer, ankyrinG, antagonizes receptor endocytosis in the AIS, causes receptor accumulation in the AIS, and leads to polarity deficits with subsequent morphological and behavioural defects. Therefore, endocytic removal of polarized receptors that diffuse into the AIS serves as a membrane-clearance mechanism that is likely to work in conjunction with the known AIS diffusion-barrier mechanism to maintain neuronal polarity on the plasma membrane. Our results reveal a conserved endocytic clearance mechanism in the AIS to maintain neuronal polarity by reinforcing axonal and dendritic compartment membrane boundaries.
Nathaniel W Mabe, Min Huang, Guillermo N Dalton, Gabriela Alexe, Daniel A Schaefer, Anna C Geraghty, Amanda L Robichaud, Amy S Conway, Delan Khalid, Marius M Mader, Julia A Belk, Kenneth N Ross, Michal Sheffer, Miles H Linde, Nghi Ly, Winnie Yao, Maria Caterina Rotiroti, Benjamin A H Smith, Marius Wernig, Carolyn R Bertozzi, Michelle Monje, Constantine S Mitsiades, Ravindra Majeti, Ansuman T Satpathy, Kimberly Stegmaier, Robbie G Majzner
Immunotherapy with anti-GD2 antibodies has advanced the treatment of children with high-risk neuroblastoma, but nearly half of patients relapse, and little is known about mechanisms of resistance to anti-GD2 therapy. Here, we show that reduced GD2 expression was significantly correlated with the mesenchymal cell state in neuroblastoma and that a forced adrenergic-to-mesenchymal transition (AMT) conferred downregulation of GD2 and resistance to anti-GD2 antibody. Mechanistically, low-GD2-expressing cell lines demonstrated significantly reduced expression of the ganglioside synthesis enzyme ST8SIA1 (GD3 synthase), resulting in a bottlenecking of GD2 synthesis. Pharmacologic inhibition of EZH2 resulted in epigenetic rewiring of mesenchymal neuroblastoma cells and re-expression of ST8SIA1, restoring surface expression of GD2 and sensitivity to anti-GD2 antibody. These data identify developmental lineage as a key determinant of sensitivity to anti-GD2 based immunotherapies and credential EZH2 inhibitors for clinical testing in combination with anti-GD2 antibody to enhance outcomes for children with neuroblastoma.
Immunotherapy with anti-GD2 antibodies has advanced the treatment of children with high-risk neuroblastoma, but nearly half of patients relapse, and little is known about mechanisms of resistance to anti-GD2 therapy. Here, we show that reduced GD2 expression was significantly correlated with the mesenchymal cell state in neuroblastoma and that a forced adrenergic-to-mesenchymal transition (AMT) conferred downregulation of GD2 and resistance to anti-GD2 antibody. Mechanistically, low-GD2-expressing cell lines demonstrated significantly reduced expression of the ganglioside synthesis enzyme ST8SIA1 (GD3 synthase), resulting in a bottlenecking of GD2 synthesis. Pharmacologic inhibition of EZH2 resulted in epigenetic rewiring of mesenchymal neuroblastoma cells and re-expression of ST8SIA1, restoring surface expression of GD2 and sensitivity to anti-GD2 antibody. These data identify developmental lineage as a key determinant of sensitivity to anti-GD2 based immunotherapies and credential EZH2 inhibitors for clinical testing in combination with anti-GD2 antibody to enhance outcomes for children with neuroblastoma.
Koji Tanabe, Hiroko Nobuta, Nan Yang, Cheen Euong Ang, Philip Huie, Sacha Jordan, Michael C Oldham, David H Rowitch, Marius Wernig
Oligodendrocytes, the myelinating cells of the central nervous system, possess great potential for disease modeling and cell transplantation-based therapies for leukodystrophies. However, caveats to oligodendrocyte differentiation protocols ( Ehrlich et al., 2017; Wang et al., 2013; Douvaras and Fossati, 2015) from human embryonic stem and induced pluripotent stem cells (iPSCs), which include slow and inefficient differentiation, and tumorigenic potential of contaminating undifferentiated pluripotent cells, are major bottlenecks towards their translational utility. Here, we report the rapid generation of human oligodendrocytes by direct lineage conversion of human dermal fibroblasts (HDFs). We show that the combination of the four transcription factors OLIG2, SOX10, ASCL1 and NKX2.2 is sufficient to convert HDFs to induced oligodendrocyte precursor cells (iOPCs). iOPCs resemble human primary and iPSC-derived OPCs based on morphology and transcriptomic analysis. Importantly, iOPCs can differentiate into mature myelinating oligodendrocytes in vitro and in vivo. Finally, iOPCs derived from patients with Pelizaeus Merzbacher disease, a hypomyelinating leukodystrophy caused by mutations in the proteolipid protein 1 (PLP1) gene, showed increased cell death compared with iOPCs from healthy donors. Thus, human iOPCs generated by direct lineage conversion represent an attractive new source for human cell-based disease models and potentially myelinating cell grafts.
Oligodendrocytes, the myelinating cells of the central nervous system, possess great potential for disease modeling and cell transplantation-based therapies for leukodystrophies. However, caveats to oligodendrocyte differentiation protocols ( Ehrlich et al., 2017; Wang et al., 2013; Douvaras and Fossati, 2015) from human embryonic stem and induced pluripotent stem cells (iPSCs), which include slow and inefficient differentiation, and tumorigenic potential of contaminating undifferentiated pluripotent cells, are major bottlenecks towards their translational utility. Here, we report the rapid generation of human oligodendrocytes by direct lineage conversion of human dermal fibroblasts (HDFs). We show that the combination of the four transcription factors OLIG2, SOX10, ASCL1 and NKX2.2 is sufficient to convert HDFs to induced oligodendrocyte precursor cells (iOPCs). iOPCs resemble human primary and iPSC-derived OPCs based on morphology and transcriptomic analysis. Importantly, iOPCs can differentiate into mature myelinating oligodendrocytes in vitro and in vivo. Finally, iOPCs derived from patients with Pelizaeus Merzbacher disease, a hypomyelinating leukodystrophy caused by mutations in the proteolipid protein 1 (PLP1) gene, showed increased cell death compared with iOPCs from healthy donors. Thus, human iOPCs generated by direct lineage conversion represent an attractive new source for human cell-based disease models and potentially myelinating cell grafts.
Markus Wöhr, Wendy M Fong, Justyna A Janas, Moritz Mall, Christian Thome, Madhuri Vangipuram, Lingjun Meng, Thomas C Südhof, Marius Wernig
BACKGROUND: The zinc finger domain containing transcription factor Myt1l is tightly associated with neuronal identity and is the only transcription factor known that is both neuron-specific and expressed in all neuronal subtypes. We identified Myt1l as a powerful reprogramming factor that, in combination with the proneural bHLH factor Ascl1, could induce neuronal fate in fibroblasts. Molecularly, we found it to repress many non-neuronal gene programs, explaining its supportive role to induce and safeguard neuronal identity in combination with proneural bHLH transcriptional activators. Moreover, human genetics studies found MYT1L mutations to cause intellectual disability and autism spectrum disorder often coupled with obesity. METHODS: Here, we generated and characterized Myt1l-deficient mice. A comprehensive, longitudinal behavioral phenotyping approach was applied. RESULTS: Myt1l was necessary for survival beyond 24 h but not for overall histological brain organization. Myt1l heterozygous mice became increasingly overweight and exhibited multifaceted behavioral alterations. In mouse pups, Myt1l haploinsufficiency caused mild alterations in early socio-affective communication through ultrasonic vocalizations. In adulthood, Myt1l heterozygous mice displayed hyperactivity due to impaired habituation learning. Motor performance was reduced in Myt1l heterozygous mice despite intact motor learning, possibly due to muscular hypotonia. While anxiety-related behavior was reduced, acoustic startle reactivity was enhanced, in line with higher sensitivity to loud sound. Finally, Myt1l haploinsufficiency had a negative impact on contextual fear memory retrieval, while cued fear memory retrieval appeared to be intact. LIMITATIONS: In future studies, additional phenotypes might be identified and a detailed characterization of direct reciprocal social interaction behavior might help to reveal effects of Myt1l haploinsufficiency on social behavior in juvenile and adult mice. CONCLUSIONS: Behavioral alterations in Myt1l haploinsufficient mice recapitulate several clinical phenotypes observed in humans carrying heterozygous MYT1L mutations and thus serve as an informative model of the human MYT1L syndrome.
BACKGROUND: The zinc finger domain containing transcription factor Myt1l is tightly associated with neuronal identity and is the only transcription factor known that is both neuron-specific and expressed in all neuronal subtypes. We identified Myt1l as a powerful reprogramming factor that, in combination with the proneural bHLH factor Ascl1, could induce neuronal fate in fibroblasts. Molecularly, we found it to repress many non-neuronal gene programs, explaining its supportive role to induce and safeguard neuronal identity in combination with proneural bHLH transcriptional activators. Moreover, human genetics studies found MYT1L mutations to cause intellectual disability and autism spectrum disorder often coupled with obesity. METHODS: Here, we generated and characterized Myt1l-deficient mice. A comprehensive, longitudinal behavioral phenotyping approach was applied. RESULTS: Myt1l was necessary for survival beyond 24 h but not for overall histological brain organization. Myt1l heterozygous mice became increasingly overweight and exhibited multifaceted behavioral alterations. In mouse pups, Myt1l haploinsufficiency caused mild alterations in early socio-affective communication through ultrasonic vocalizations. In adulthood, Myt1l heterozygous mice displayed hyperactivity due to impaired habituation learning. Motor performance was reduced in Myt1l heterozygous mice despite intact motor learning, possibly due to muscular hypotonia. While anxiety-related behavior was reduced, acoustic startle reactivity was enhanced, in line with higher sensitivity to loud sound. Finally, Myt1l haploinsufficiency had a negative impact on contextual fear memory retrieval, while cued fear memory retrieval appeared to be intact. LIMITATIONS: In future studies, additional phenotypes might be identified and a detailed characterization of direct reciprocal social interaction behavior might help to reveal effects of Myt1l haploinsufficiency on social behavior in juvenile and adult mice. CONCLUSIONS: Behavioral alterations in Myt1l haploinsufficient mice recapitulate several clinical phenotypes observed in humans carrying heterozygous MYT1L mutations and thus serve as an informative model of the human MYT1L syndrome.
Friederike Matheus, Tal Raveh, Anthony E Oro, Marius Wernig, Micha Drukker
The manipulation of human leukocyte antigens (HLAs) and immune modulatory factors in "universal" human pluripotent stem cells (PSCs) holds promise for immunological tolerance without HLA matching. This paradigm raises concerns should "universal" grafts become virally infected. Furthermore, immunological manipulation might functionally impair certain progeny, such as hematopoietic stem cells. We discuss the risks and benefits of hypoimmunogenic PSCs, and the need to further advance HLA matching and autologous strategies.
The manipulation of human leukocyte antigens (HLAs) and immune modulatory factors in "universal" human pluripotent stem cells (PSCs) holds promise for immunological tolerance without HLA matching. This paradigm raises concerns should "universal" grafts become virally infected. Furthermore, immunological manipulation might functionally impair certain progeny, such as hematopoietic stem cells. We discuss the risks and benefits of hypoimmunogenic PSCs, and the need to further advance HLA matching and autologous strategies.
Yohei Shibuya, Kevin K Kumar, Marius Marc-Daniel Mader, Yongjin Yoo, Luis Angel Ayala, Mu Zhou, Manuel Alexander Mohr, Gernot Neumayer, Ishan Kumar, Ryo Yamamoto, Paul Marcoux, Benjamin Liou, F Chris Bennett, Hiromitsu Nakauchi, Ying Sun, Xiaoke Chen, Frank L Heppner, Tony Wyss-Coray, Thomas C Südhof, Marius Wernig
Hematopoietic cell transplantation after myeloablative conditioning has been used to treat various genetic metabolic syndromes but is largely ineffective in diseases affecting the brain presumably due to poor and variable myeloid cell incorporation into the central nervous system. Here, we developed and characterized a near-complete and homogeneous replacement of microglia with bone marrow cells in mice without the need for genetic manipulation of donor or host. The high chimerism resulted from a competitive advantage of scarce donor cells during microglia repopulation rather than enhanced recruitment from the periphery. Hematopoietic stem cells, but not immediate myeloid or monocyte progenitor cells, contained full microglia replacement potency equivalent to whole bone marrow. To explore its therapeutic potential, we applied microglia replacement to a mouse model for Prosaposin deficiency, which is characterized by a progressive neurodegeneration phenotype. We found a reduction of cerebellar neurodegeneration and gliosis in treated brains, improvement of motor and balance impairment, and life span extension even with treatment started in young adulthood. This proof-of-concept study suggests that efficient microglia replacement may have therapeutic efficacy for a variety of neurological diseases.
Hematopoietic cell transplantation after myeloablative conditioning has been used to treat various genetic metabolic syndromes but is largely ineffective in diseases affecting the brain presumably due to poor and variable myeloid cell incorporation into the central nervous system. Here, we developed and characterized a near-complete and homogeneous replacement of microglia with bone marrow cells in mice without the need for genetic manipulation of donor or host. The high chimerism resulted from a competitive advantage of scarce donor cells during microglia repopulation rather than enhanced recruitment from the periphery. Hematopoietic stem cells, but not immediate myeloid or monocyte progenitor cells, contained full microglia replacement potency equivalent to whole bone marrow. To explore its therapeutic potential, we applied microglia replacement to a mouse model for Prosaposin deficiency, which is characterized by a progressive neurodegeneration phenotype. We found a reduction of cerebellar neurodegeneration and gliosis in treated brains, improvement of motor and balance impairment, and life span extension even with treatment started in young adulthood. This proof-of-concept study suggests that efficient microglia replacement may have therapeutic efficacy for a variety of neurological diseases.
Jie Wang, Yi Miao, Rebecca Wicklein, Zijun Sun, Jinzhao Wang, Kevin M Jude, Ricardo A Fernandes, Sean A Merrill, Marius Wernig, K Christopher Garcia, Thomas C Südhof
RTN4-binding proteins were widely studied as "NoGo" receptors, but their physiological interactors and roles remain elusive. Similarly, BAI adhesion-GPCRs were associated with numerous activities, but their ligands and functions remain unclear. Using unbiased approaches, we observed an unexpected convergence: RTN4 receptors are high-affinity ligands for BAI adhesion-GPCRs. A single thrombospondin type 1-repeat (TSR) domain of BAIs binds to the leucine-rich repeat domain of all three RTN4-receptor isoforms with nanomolar affinity. In the 1.65 Å crystal structure of the BAI1/RTN4-receptor complex, C-mannosylation of tryptophan and O-fucosylation of threonine in the BAI TSR-domains creates a RTN4-receptor/BAI interface shaped by unusual glycoconjugates that enables high-affinity interactions. In human neurons, RTN4 receptors regulate dendritic arborization, axonal elongation, and synapse formation by differential binding to glial versus neuronal BAIs, thereby controlling neural network activity. Thus, BAI binding to RTN4/NoGo receptors represents a receptor-ligand axis that, enabled by rare post-translational modifications, controls development of synaptic circuits.
RTN4-binding proteins were widely studied as "NoGo" receptors, but their physiological interactors and roles remain elusive. Similarly, BAI adhesion-GPCRs were associated with numerous activities, but their ligands and functions remain unclear. Using unbiased approaches, we observed an unexpected convergence: RTN4 receptors are high-affinity ligands for BAI adhesion-GPCRs. A single thrombospondin type 1-repeat (TSR) domain of BAIs binds to the leucine-rich repeat domain of all three RTN4-receptor isoforms with nanomolar affinity. In the 1.65 Å crystal structure of the BAI1/RTN4-receptor complex, C-mannosylation of tryptophan and O-fucosylation of threonine in the BAI TSR-domains creates a RTN4-receptor/BAI interface shaped by unusual glycoconjugates that enables high-affinity interactions. In human neurons, RTN4 receptors regulate dendritic arborization, axonal elongation, and synapse formation by differential binding to glial versus neuronal BAIs, thereby controlling neural network activity. Thus, BAI binding to RTN4/NoGo receptors represents a receptor-ligand axis that, enabled by rare post-translational modifications, controls development of synaptic circuits.
Daniel D Lam, Rhîannan H Williams, Ernesto Lujan, Koji Tanabe, Georg Huber, Nay Lui Saw, Juliane Merl-Pham, Aaro V Salminen, David Lohse, Sally Spendiff, Melanie J Plastini, Michael Zech, Hanns Lochmüller, Arie Geerlof, Stefanie M Hauck, Mehrdad Shamloo, Marius Wernig, Juliane Winkelmann
Collagen VI is a key component of muscle basement membranes, and genetic variants can cause monogenic muscular dystrophies. Conversely, human genetic studies recently implicated collagen VI in central nervous system function, with variants causing the movement disorder dystonia. To elucidate the neurophysiological role of collagen VI, we generated mice with a truncation of the dystonia-related collagen α3 (VI) (COL6A3) C-terminal domain (CTD). These Col6a3 CTT mice showed a recessive dystonia-like phenotype in both sexes. We found that COL6A3 interacts with the cannabinoid receptor 1 (CB1R) complex in a CTD-dependent manner. Col6a3 CTT mice of both sexes have impaired homeostasis of excitatory input to the basal pontine nuclei (BPN), a motor control hub with dense COL6A3 expression, consistent with deficient endocannabinoid signaling. Aberrant synaptic input in the BPN was normalized by a CB1R agonist, and motor performance in Col6a3 CTT mice of both sexes was improved by CB1R agonist treatment. Our findings identify a readily therapeutically addressable synaptic mechanism for motor control.SIGNIFICANCE STATEMENT Dystonia is a movement disorder characterized by involuntary movements. We previously identified genetic variants affecting a specific domain of the COL6A3 protein as a cause of dystonia. Here, we created mice lacking the affected domain and observed an analogous movement disorder. Using a protein interaction screen, we found that the affected COL6A3 domain mediates an interaction with the cannabinoid receptor CB1R. Concordantly, our COL6A3-deficient mice showed a deficit in synaptic plasticity linked to a deficit in cannabinoid signaling. Pharmacological cannabinoid augmentation rescued the motor impairment of the mice. Thus, cannabinoid augmentation could be a promising avenue for treating dystonia, and we have identified a possible molecular mechanism mediating this.
Collagen VI is a key component of muscle basement membranes, and genetic variants can cause monogenic muscular dystrophies. Conversely, human genetic studies recently implicated collagen VI in central nervous system function, with variants causing the movement disorder dystonia. To elucidate the neurophysiological role of collagen VI, we generated mice with a truncation of the dystonia-related collagen α3 (VI) (COL6A3) C-terminal domain (CTD). These Col6a3 CTT mice showed a recessive dystonia-like phenotype in both sexes. We found that COL6A3 interacts with the cannabinoid receptor 1 (CB1R) complex in a CTD-dependent manner. Col6a3 CTT mice of both sexes have impaired homeostasis of excitatory input to the basal pontine nuclei (BPN), a motor control hub with dense COL6A3 expression, consistent with deficient endocannabinoid signaling. Aberrant synaptic input in the BPN was normalized by a CB1R agonist, and motor performance in Col6a3 CTT mice of both sexes was improved by CB1R agonist treatment. Our findings identify a readily therapeutically addressable synaptic mechanism for motor control.SIGNIFICANCE STATEMENT Dystonia is a movement disorder characterized by involuntary movements. We previously identified genetic variants affecting a specific domain of the COL6A3 protein as a cause of dystonia. Here, we created mice lacking the affected domain and observed an analogous movement disorder. Using a protein interaction screen, we found that the affected COL6A3 domain mediates an interaction with the cannabinoid receptor CB1R. Concordantly, our COL6A3-deficient mice showed a deficit in synaptic plasticity linked to a deficit in cannabinoid signaling. Pharmacological cannabinoid augmentation rescued the motor impairment of the mice. Thus, cannabinoid augmentation could be a promising avenue for treating dystonia, and we have identified a possible molecular mechanism mediating this.
Hannah Shelby, Tara Shelby, Marius Wernig
Embryonic development and cell specification have been viewed as an epigenetically rigid process. Through accumulation of irreversible epigenetic marks, the differentiation process has been considered unidirectional, and once completed cell specification would be permanent and stable. However, somatic cell nuclear transfer that involved the implantation of a somatic nucleus into a previously enucleated oocyte accomplished in amphibians in the 1950s and in mammals in the late 1990s-resulting in the birth of "Dolly the sheep"-clearly showed that "terminal" differentiation is reversible. In parallel, work on lineage-determining factors like MyoD revealed surprising potential to modulate lineage identity in somatic cells. This work culminated in the discovery that a set of four defined factors can reprogram fibroblasts into induced pluripotent stem (iPS) cells, which were shown to be molecularly and functionally equivalent to blastocyst-derived embryonic stem (ES) cells, thus essentially showing that defined factors can induce authentic reprogramming without the need of oocytes. This concept was further extended when it was shown that fibroblasts can be directly converted into neurons, showing induced lineage conversion is possible even between cells representing two different germ layers. These findings suggest that "everything is possible" (i.e., once key lineage reprogramming factors are identified, cells should be able to convert into any desired lineage).
Embryonic development and cell specification have been viewed as an epigenetically rigid process. Through accumulation of irreversible epigenetic marks, the differentiation process has been considered unidirectional, and once completed cell specification would be permanent and stable. However, somatic cell nuclear transfer that involved the implantation of a somatic nucleus into a previously enucleated oocyte accomplished in amphibians in the 1950s and in mammals in the late 1990s-resulting in the birth of "Dolly the sheep"-clearly showed that "terminal" differentiation is reversible. In parallel, work on lineage-determining factors like MyoD revealed surprising potential to modulate lineage identity in somatic cells. This work culminated in the discovery that a set of four defined factors can reprogram fibroblasts into induced pluripotent stem (iPS) cells, which were shown to be molecularly and functionally equivalent to blastocyst-derived embryonic stem (ES) cells, thus essentially showing that defined factors can induce authentic reprogramming without the need of oocytes. This concept was further extended when it was shown that fibroblasts can be directly converted into neurons, showing induced lineage conversion is possible even between cells representing two different germ layers. These findings suggest that "everything is possible" (i.e., once key lineage reprogramming factors are identified, cells should be able to convert into any desired lineage).
Jie Wang, Yi Miao, Rebecca Wicklein, Zijun Sun, Jinzhao Wang, Kevin M Jude, Ricardo A Fernandes, Sean A Merrill, Marius Wernig, K Christopher Garcia, Thomas C Südhof
RTN4-binding proteins were widely studied as "NoGo" receptors, but their physiological interactors and roles remain elusive. Similarly, BAI adhesion-GPCRs were associated with numerous activities, but their ligands and functions remain unclear. Using unbiased approaches, we observed an unexpected convergence: RTN4 receptors are high-affinity ligands for BAI adhesion-GPCRs. A single thrombospondin type 1-repeat (TSR) domain of BAIs binds to the leucine-rich repeat domain of all three RTN4-receptor isoforms with nanomolar affinity. In the 1.65 Å crystal structure of the BAI1/RTN4-receptor complex, C-mannosylation of tryptophan and O-fucosylation of threonine in the BAI TSR-domains creates a RTN4-receptor/BAI interface shaped by unusual glycoconjugates that enables high-affinity interactions. In human neurons, RTN4 receptors regulate dendritic arborization, axonal elongation, and synapse formation by differential binding to glial versus neuronal BAIs, thereby controlling neural network activity. Thus, BAI binding to RTN4/NoGo receptors represents a receptor-ligand axis that, enabled by rare post-translational modifications, controls development of synaptic circuits.
RTN4-binding proteins were widely studied as "NoGo" receptors, but their physiological interactors and roles remain elusive. Similarly, BAI adhesion-GPCRs were associated with numerous activities, but their ligands and functions remain unclear. Using unbiased approaches, we observed an unexpected convergence: RTN4 receptors are high-affinity ligands for BAI adhesion-GPCRs. A single thrombospondin type 1-repeat (TSR) domain of BAIs binds to the leucine-rich repeat domain of all three RTN4-receptor isoforms with nanomolar affinity. In the 1.65 Å crystal structure of the BAI1/RTN4-receptor complex, C-mannosylation of tryptophan and O-fucosylation of threonine in the BAI TSR-domains creates a RTN4-receptor/BAI interface shaped by unusual glycoconjugates that enables high-affinity interactions. In human neurons, RTN4 receptors regulate dendritic arborization, axonal elongation, and synapse formation by differential binding to glial versus neuronal BAIs, thereby controlling neural network activity. Thus, BAI binding to RTN4/NoGo receptors represents a receptor-ligand axis that, enabled by rare post-translational modifications, controls development of synaptic circuits.
Yi Han Ng, Soham Chanda, Justyna A Janas, Nan Yang, Yuko Kokubu, Thomas C Südhof, Marius Wernig
The differentiation of pluripotent stem cells can be accomplished by sequential activation of signaling pathways or through transcription factor programming. Multistep differentiation imitates embryonic development to obtain authentic cell types, but it suffers from asynchronous differentiation with variable efficiency. Transcription factor programming induces synchronous and efficient differentiation with higher reproducibility but may not always yield authentic cell types. We systematically...
The differentiation of pluripotent stem cells can be accomplished by sequential activation of signaling pathways or through transcription factor programming. Multistep differentiation imitates embryonic development to obtain authentic cell types, but it suffers from asynchronous differentiation with variable efficiency. Transcription factor programming induces synchronous and efficient differentiation with higher reproducibility but may not always yield authentic cell types. We systematically...
Vierbuchen T, Wernig M
Classic experiments such as somatic cell nuclear transfer into oocytes and cell fusion demonstrated that differentiated cells are not irreversibly committed to their fate. More recent work has built on these conclusions and discovered defined factors that directly induce one specific cell type from another, which may be as distantly related as cells from different germ layers. This suggests the possibility that any specific cell type may be directly converted into any other if the appropriate reprogramming factors are known. Direct lineage conversion could provide important new sources of human cells for modeling disease processes or for cellular-replacement therapies. For future applications, it will be critical to carefully determine the fidelity of reprogramming and to develop methods for robustly and efficiently generating human cell types of interest.
Classic experiments such as somatic cell nuclear transfer into oocytes and cell fusion demonstrated that differentiated cells are not irreversibly committed to their fate. More recent work has built on these conclusions and discovered defined factors that directly induce one specific cell type from another, which may be as distantly related as cells from different germ layers. This suggests the possibility that any specific cell type may be directly converted into any other if the appropriate reprogramming factors are known. Direct lineage conversion could provide important new sources of human cells for modeling disease processes or for cellular-replacement therapies. For future applications, it will be critical to carefully determine the fidelity of reprogramming and to develop methods for robustly and efficiently generating human cell types of interest.
Daniel Haag, Norman Mack, Patricia Benites Goncalves da Silva, Britta Statz, Jessica Clark, Koji Tanabe, Tanvi Sharma, Natalie Jäger, David T W Jones, Daisuke Kawauchi, Marius Wernig, Stefan M Pfister
Diffuse intrinsic pontine glioma (DIPG) is an aggressive childhood tumor of the brainstem with currently no curative treatment available. The vast majority of DIPGs carry a histone H3 mutation leading to a lysine 27-to-methionine exchange (H3K27M). We engineered human induced pluripotent stem cells (iPSCs) to carry an inducible H3.3-K27M allele in the endogenous locus and studied the effects of the mutation in different disease-relevant neural cell types. H3.3-K27M upregulated bivalent promoter-associated developmental genes, producing diverse outcomes in different cell types. While being fatal for iPSCs, H3.3-K27M increased proliferation in neural stem cells (NSCs) and to a lesser extent in oligodendrocyte progenitor cells (OPCs). Only NSCs gave rise to tumors upon induction of H3.3-K27M and TP53 inactivation in an orthotopic xenograft model recapitulating human DIPGs. In NSCs, H3.3-K27M leads to maintained expression of stemness and proliferative genes and a premature activation of OPC programs that together may cause tumor initiation.
Diffuse intrinsic pontine glioma (DIPG) is an aggressive childhood tumor of the brainstem with currently no curative treatment available. The vast majority of DIPGs carry a histone H3 mutation leading to a lysine 27-to-methionine exchange (H3K27M). We engineered human induced pluripotent stem cells (iPSCs) to carry an inducible H3.3-K27M allele in the endogenous locus and studied the effects of the mutation in different disease-relevant neural cell types. H3.3-K27M upregulated bivalent promoter-associated developmental genes, producing diverse outcomes in different cell types. While being fatal for iPSCs, H3.3-K27M increased proliferation in neural stem cells (NSCs) and to a lesser extent in oligodendrocyte progenitor cells (OPCs). Only NSCs gave rise to tumors upon induction of H3.3-K27M and TP53 inactivation in an orthotopic xenograft model recapitulating human DIPGs. In NSCs, H3.3-K27M leads to maintained expression of stemness and proliferative genes and a premature activation of OPC programs that together may cause tumor initiation.
Marius Wernig
wernig@stanford.edu
Dr. Marius Wernig is a Professor of Pathology and a Co-Director of the Institute for Stem Cell Biology and Regenerative Medicine at Stanford University. He graduated with an M.D. Ph.D. from the Technical University of Munich where he trained in developmental genetics in the lab of Rudi Balling. After completing his residency in Neuropathology and General Pathology at the University of Bonn, he then became a postdoctoral fellow in the lab of Dr. Rudolf Jaenisch at the Whitehead Institute for Biomedical Research/ MIT in Cambridge, MA. In 2008, Dr. Wernig joined the faculty of the Institute for Stem Cell Biology and Regenerative Medicine at Stanford University where he has been ever since.
He received an NIH Pathway to Independence Award, the Cozzarelli Prize for Outstanding Scientific Excellence from the National Academy of Sciences U.S.A., the Outstanding Investigator Award from the International Society for Stem Cell Research, the New York Stem Cell Foundation Robertson Stem Cell Prize, the Ogawa-Yamanaka Stem Cell Prize delivered by the Gladstone Institute and more recently has been named a Faculty Scholar by the Howard Hughes Medical Institute.
Dr. Wernig’s lab is interested in pluripotent stem cell biology and the molecular determinants of neural cell fate decisions. His laboratory was the first to generate functional neuronal cells reprogrammed directly from skin fibroblasts, which he termed induced neuronal (iN) cells. The lab is now working on identifying the molecular mechanisms underlying induced lineage fate changes, the phenotypic consequences of disease-causing mutations in human neurons and other neural lineages as well as the development of novel therapeutic gene targeting and cell transplantation-based strategies for a variety of monogenetic diseases.
Academic appointments
Associate Professor Institute for Stem Cell Biology and Regenerative Medicine
Member:
Bio-X
Cardiovascular Institute
Child Health Research Institute
Institute for Stem Cell Biology and Regenerative Medicine
Stanford Cancer Institute
Stanford Neurosciences Institute
Administrative appointments
Faculty Senate, Department of Pathology (2017 - Present)
Assistant Professor, Institute for Stem Cell Biology and Regenerative Medicine (2008 - 2014)
Honors & Awards
HHMI Faculty Scholar Award, Howard Hughes Medical Institute (2016)
New York Stem Cell Foundation Robertson Stem Cell Prize, New York Stem Cell Foundation (2014)
The Outstanding Young Investigator Award, International Society for Stem Cell Research (2013)
Ascina Award, Republic of Austria (2010)
Cozzarelli Prize for Outstanding Scientific Excellence, National Academy of Sciences USA (2009)
New Scholar in Aging, Ellison Medical Foundation (2010)
Robertson Investigator Award, New York Stem Cell Foundation (2010)
Donald E. and Delia B. Baxter Faculty Scholarship, Stanford University (2009)
Margaret and Herman Sokol Award, Biomedical Research (2007)
Longterm fellowship Human Frontiers Science Program Organisation, HFSP (2004-2006)
Boards, Advisory Committees
Professional Organizations Member, Society for Neuroscience (2003 - Present)
Member, International Society for Stem Cell Research (2004 - Present)
Editorial Board Member, Cell Stem Cell (2012 - Present)
Editorial Board Member, Stem Cell Reports (2013 - Present)
Member, Program Committee, Society for Neuroscience (2016 - Present)
Chair, Program Committee, International Society for Stem Cell Research (2017 - Present)
Professional Education
M.D., Technical University of Munich, Medicine (2000)
Team

I joined the Wernig lab as an LSRP in the summer of 2022 after graduating from UCLA. I’m interested in the regenerative capabilities of the human body and the molecular mechanisms underlying these regenerative systems, as well as the ways in which we can utilize these mechanisms to treat disease via tools such as cell and gene therapy. My research interests further include immunological topics such as cancer biology and the coevolution of pathogenic species and the immune response. In the Wernig lab, I am investigating methods of replacing dysfunctional microglia in the CNS in the context of neurodegenerative diseases, as well as the translational potential of gene editing and the role that genetically manipulated cells can play in treating such diseases. Outside the lab, I enjoy spending time at the beach, watching sports, and going on road trips with my friends.
Danwei Wu is a Stanford neurology resident in the Neuroscience Scholar Track and aspiring neuroimmunologist. Prior to starting residency, she completed an HHMI Medical Research Fellowship with Dr. Vann Bennett at Duke University studying neuron-specific membrane domains and their interaction with cytoskeletal structures. Her current research interest includes cell-based therapies for multiple sclerosis, molecular pathways of neuro-repair, and pathogenesis of autoimmunity. She is interested in developing new therapies for neurologic diseases. Outside of the lab, she enjoys hiking and reading science fiction.
I am a PhD student in the Neurosciences Interdepartmental Program and joined the lab in summer 2022. I am interested in developing methods to reprogram stem cells directly into oligodendrocytes. I then hope to use these cells to study the role of oligodendrocytes in aging and neurodegeneration. In my free time I like to read, ski, and play tennis.
I am a passionate cellular and molecular biologistwith expertise in research related to cancer, genomic/chromosomal instability, DNA damage response, epigenetics, proteomics and cellular identity. The latter topic attracted me to the Wernig lab where I aim to decipher the mechanisms that allow us to specifically switch the identity of a cell, converting differentiated somatic cells into induced neurons. I also work on a project that aims to integrate CRISPR/CAS9-mediated gene correction with iPS cell generation in order to establish a therapy for the devastating skin disease epidermolysis bullosa. In my free time I play underwater rugby, surf, spearfish and ski!
I am an undergraduate studying neurobiology and computer science. In the Wernig Lab, I am studying how the interactions between reprogramming factors and chromatin modifiers allow for fully developed fibrojacklynl@stanford.edublasts to reprogram into induced neuronal cells (iN).
I studied small GTPase signaling and how its perturbation can contribute to cancer and neurological disorders during my PhD training at Cold Spring Harbor Laboratory. Since then, I have become increasingly fascinated by the epigenetic mechanisms of gene regulation, and how changes in epigenome influence cell function and fate decisions. In the Wernig lab, I am currently investigating the interactions between the reprogramming factors and chromatin modifiers. More specifically, I am interested in finding out how such interactions enable a terminally differentiated cell—for example, a fibroblast—to acquire new transcriptional program that allows its reprogramming into induced neuron (iN).
I am a Bachelor of Science candidate in Bioengineering at Stanford University. My research in the Wernig Lag involves investigating the relationship between microglia and the IKBKAP gene, specifically its role in mechanisms such as microglia regeneration and the transdifferentiation of hematopoietic stem and progenitor cells into microglia-like cells.
Kayla is a PhD student in the Neurosciences Interdepartmental Program and joined the lab in summer 2022. She is interested in leveraging the lab's cell reprogramming techniques to study the roles of individual cell types in proteinopathies. She aims to faithfully recapitulate disease states and uncover the underlying molecular mechanisms that lead to neurodegeneration. In her free time, Kayla can be found birding, hiking, or performing with the Stanford Symphony Orchestra.
I received my BA in molecular and cell biology (Neurobiology focus) and computer science from UC Berkeley. I went on to do my PhD at Duke University, where I worked on engineering electrical synapses for precise neuromodulation in Kafui Dzirasa’s lab. Now, as a postdoc in Marius Wernig lab, I am interested in delving deeper into cellular engineering to better understand neuronal induction, synapse formation, and deliver treatment across the blood brain barrier using engineered microglia. In my free time, I enjoy cooking and coding/electrical engineering projects.
As a neurosurgical resident, my research interests include translational topics such as cell therapy and neuroregenerative mechanisms. In the Wernig lab, I study the cellular reprogramming potential of microglia. Moreover, I’m interested in the functional integration of induced neurons into neural systems.
I am an undergraduate student studying Human Biology. At Wernig Lab, I am helping to research how microglial cells organize and communicate with each other. I am interested in understanding the relationship between changes in the organization of microglia and other neural cells and the progression of neuronal diseases.
I received my Ph.D. in Neuroscience and Brain Technologies in 2021 from the University of Genoa and the Italian Institute of Technology, Italy. I joined the Wernig lab in December 2022. Here, I am interested in understanding how the microglia-derived factors end up in the neuronal lysosomes. Furthermore, I am interested in understanding how the accumulation and breakdown of these factors lead to neurodegenerative disorders using human induced pluripotent stem cell culture. I read non-fiction novels, sketch, and go for hikes in my free time.
Rida Rehman received her doctorate in Natural Sciences (Dr. Rer. Nat.), specializing in brain trauma and rehabilitation at Universtät Ulm, Germany in 2023. She also holds an M.S. in Biomedical Sciences and a B.S. in Applied Biosciences from NUST, Pakistan. Rida’s research has primarily focused on understanding the critical role of microglia in neurotrauma, degeneration, reprogramming and repair. Rida joined Wernig lab in 2023 and is primarily working on microglia to neuron reprogramming.
I worked as a neurologist for 10 years in Japan. During clinical practice, I saw a lot of patients who could not be treated by the current medicine. As a result, I became strongly attracted to basic research of neuroscience. I received my PhD in Dr. Tatsushi Toda’s lab at Kobe University, where I studied disease-modifying drug for Parkinson’s disease. As a post doc fellow in the Wernig lab, I’m interested in investigating the ubiquitylation enzymes that are associated with the pathophysiology of Alzheimer’s disease in order to cure patients with neurodegenerative diseases in the future. In my free time, I play with my children, go cycling, and read comics.
I am a PhD student in the Department of Neurosciences and I joined the Wernig lab in the summer of 2020. I hope to study the fundamental cell biology of microglia giving rise to stable tiling in the brain. Furthermore, I am interested in how these mechanisms contribute to brain homeostasis and change with neuronal disease.
During my PhD in Yongjuan Zhao and Honcheung Lee’s lab, I focused on studying the enzymatic activity and function of ADP-ribosyl cyclase, particularly the activity of SARM1 and its role in axon degeneration. This work sparked my interest in neuroscience and neuropathology. Microglia, the resident immune cells in the brain, play critical roles in neuronal function and can be either beneficial or detrimental in the context of neurodegenerative diseases.In Wernig’s lab, I am interested in investigating the function of microglia in aging and Alzheimer’s disease. My current research focuses on understanding the effects of clonal hematopoietic mutations on microglia and their role in the development of Alzheimer’s disease.
I'm excited about diving into the mysteries of cell fate changes at both the cellular and molecular biology levels, using both experimental and computational tools. In the Wernig lab, I'm on a fascinating journey exploring methods to reprogramm cells into specific neuronal fates. Plus, I'm eager to unravel the basic transcriptional regulation codes in these processes.
Alumni




.gif)

Alan Napole was an LSRP working on elucidating the mechanisms of transgene silencing following iN differentiation from pluripotent stem cells. Concurrently, Alan is also working on understanding the role of microglia in multiple sclerosis via EAE mice models. Previously, he was a CIRM scholar working closely with microglia replacement strategies to develop a human mouse microglia chimera for investigation of neuroimmunological diseases. Alan is currently applying to medical school with interests in neuroscience and surgery. Outside of the lab, he enjoys attending music festivals, playing soccer, and cooking. He moved on to grad school in his home town LA. We wish him all the best for his future endeavors!
Angel earned his B.S. in Biology at California State Polytechnic University, Pomona. As an undergraduate he studied the effects of nicotine on diabetic individuals. As a CIRM fellow at Stanford University, he is interested in using microglia cell replacement and stem cell technology as a potential therapeutic for neurological disorders. Unfortunately, COVID19 cut some of the last of his time he was planning to stay with us. We are proud of Angel that he got into the PhD program at UC Irvine where he moved during these challenging times of natural disasters of the year 2020. Good luck Angel!
Bahareh's research interests were focused on epigenetic regulation of cognition at the level of synapses, using iN system. In my free time she likes to do sports and outdoor activities, cook healthy food, make mixed drinks, play music, and collect vinyl records. After graduating from our Stem Cell and Regenerative Medicine PhD program she moved on as postdoctoral fellow to Boston.
Cheen Euong Ang was a bioengineering PhD student in the Wernig lab. He spent his undergraduate career at McGill University, graduating with a B.Sc. in Chemistry with a focus in biological chemistry. Since coming to Stanford, Cheen Euong has been working on investigating the mechanisms of iN reprogramming and applying the iN platform in disease modeling and aging. Other than doing research in the lab, Cheen Euong has participated several scientific outreach programs such as the Stanford Biomedical Engineering Society undergraduate academic mentoring program and Stanford Institute of Medical Summer High School Student Research Program. Cheen moved on to do his postdoc in Xiaowei Zhuang at Harvard University in Cambridge, MA.
Christian's project focused on the function of synaptic proteins in the context of human neurons. Brain research has made tremendous advances in the last decades using animal models. However, most neuropathological conditions, such as autism or schizophrenia, are unique to human physiology. Thus, I study the effect of genetic alterations of synaptic proteins in induced human neurons using immunohistochemistry, analysis of gene and protein expression, as well as electrophysiology. Christian moved on to a senior lecturer position at the newly formed university of Linz, Austria. Good luck there!
Graduating from UC Berkeley majoring in Biology with an honors in Neuroscience, and having worked as an MD at the Royal Melbourne Hospital, I have developed a particular interest in translational neuroscience. In the lab, I am investigating the efficacy of cell therapies for neurological disorders. I am currently working on microglia based therapies for major neurological diseases, including multiple sclerosis.
I am a molecular biologist with a focus on neuro- and cancer biology. I am interested in using human iPSCs to develop new in vitro disease models, particularly for neuropsychiatric disorders and neurooncology. This led me to the Wernig lab where I started my postdoc to explore the advantages of direct neuronal reprogramming and the intricacies of step-wise differentiation of neural cell types. I am genetically engineering iPSC lines to generate complex genomic alterations and inducible mutations. Following differentiation into the corresponding disease-relevant tissue, I am puzzling together cellular phenotypes, transcriptional regulation, protein interactions, and epigenetic changes for a better understanding of the disease biology. In my spare time, I enjoy a new definition of chaos by my 2-year-old daughter.
Hiroko received Ph.D. from UCLA Neuroscience program in Jim Waschek's lab. She's currently a postdoc studying the disease mechanism of pediatric brain disorder Pelizaeus-Merzbacher Disorder, affecting oligodendrocyte development and myelination. She uses patient-derived iPS cells, gene targeting in iPS cells, and animal models.
I was an administrative associate for the Wernig Lab for a brief period in 2019. I am a native San Franciscan and I love the bay area. I received my undergrad from San Francisco State. I am fluent inSpanish so if you ever need a Spanish lesson come by desk. My husband and I have three beautiful childrenand during my time off I really enjoying spending time with my family.
I grew up in Long Island, NY. I attended college at Cornell University majoring in Biological Sciences with a concentration in Neurobiology and Behavior. After graduation in 2009, I moved to Nashville, TN to join the Medical Scientist Training Program at Vanderbilt University where I earned a combined MD/PhD. In 2016, I started my residency in Neurosurgery at Stanford. In the Wernig lab, I am interested in developing microglia-based regenerative therapies for neurodevelopmental and neurodegenerative disorders.
I received my PhD from National Institute of Biological Sciences (NIBS) at Beijing, China. My PhD mentor is Dr. Xiaodong Wang, who is a chinese-born American biochemist best known for his work with cytochrome C on apoptosis. He is a member of United States National Academy of Sciences and Howard Hughes Medical Institute.
During my PhD stage, my major work is to try and find function of a protein named RIP3 in necrosis (other program cell death) pathway. I use human fetal neural stem cells to study mutation caused epigenetic rigidity in reprogramming process. A lot of patient diseases are caused by gene mutation, but we do not know which kind of cells and which site of mutate will cause disease. We try to work on neural stem cells to induce some point mutation to imitate brain cancer disease from reported mutation site in human patient. Then we will know which mutation site is the key and try to fix it by the biology method.
The immense potential of genetic engineering technology as a tool to understand disease mechanisms and as a therapy motivates me to pursue research. I am studying CRISPR/Cas9 mediated gene correction of mutations in patients suffering from Epidermolysis bullosa. I am also studying iPSC-derived induced neurons to model neurological disorders.
Minjeong was the administrative associate for the Wernig Lab. We all loved her positive spirit that made our lab a happy place. We were fortunate to have her for a full year! During her time off, she loved hanging out with my family and trying out different recipes. She moved on to a full time position in business.
Dr. Moritz Mall is a postdoctoral fellow in the lab. After studying biochemistry and molecular biology at the LMU in Munich and the ETH in Zurich he received his Ph.D. from the EMBL in Heidelberg for his mechanistic studies on mitotic cell division. Besides surfing Moritz’s passion is to understand the molecular mechanisms of cell fate determination during reprogramming, development and disease.
A striking feature of the mammalian nervous system is its enormous cellular diversity. The biological question that drives my research in long-term is to understand how the nervous system develops and achieves its extraordinary complexity. I would also like to leverage my expertise in lineage reprogramming, stem cell biology, and neurobiology to develop reprogramming-based human cell culture models and study the fundamental processes underlying the development and function of human nervous system under normal and pathological conditions. Outside the lab, I like spending my time cycling, rock climbing and hiking.
I grew up in Australia where I completed an undergraduate degree in neuroscience and an MD at the University of Melbourne, Australia. I then completed my intern year at the Royal Melbourne Hospital. I have a keen interest in stem cell therapeutics and their potential application in treating debilitating neurological conditions. My current research focus is on novel microglia based therapies that can potentially ameliorate disease progression in multiple sclerosis.
With billions of neurons interconnected with many billions of synapses, our brain is the most complex object in the known universe. I like to think that I’m doing my part in understanding it. I use human neurons to study a synaptic protein called Neuroligin 4 that is not found in mice, therefore very complicated to study but at the same time extremely fascinating. When mutated Neuroligin 4 causes Autism and impairs the synaptic transmission of neurons. After leaving his prominent imprint on the lab he moved on to join the faculty at Mount Sanai School of Medicine in New York. Good luck Samuele, we will miss you!
My research interest is to investigate how genetic background contributes to the pathomechanism of complex mental disorders such autism or schizophrenia. In order to achieve my goals I am using various experimental techniques including rodent and human neuronal cell culture preparations, immunohistochemistry, gene and protein expression analysis and electrophysiology. As a scientist, my motto is: "We work in the dark to serve the light." Tamas moved back to his home country Hungary.
Virginia is a dentist that worked with underserved communities before taking time off to care for her family. She is happy to be the administrative associate for the Wernig lab. In her free time she enjoys reading, playing tennis, and spending time with her family. Unfortunately, due to COVID19 Virginia needed more time to care for her children and had to leave us. She was a delight to have around and we will dearly miss her!
Wendy received her Ph.D. from Columbia University, where she studied how cholesterol-rich membrane microdomains regulate a distinct function of a transmembrane protein, resulting in specific behavioral outcomes. In the Wernig lab, she aimed to understand the molecular mechanisms by which neuronal identity is established and how its disruption leads to neurological diseases focusing on the gene Myt1l.
Yingfei Liu is a Neurobiology PhD candidate from Xi`an Jiaotong University,China. She is studying here for two years as a Joint-Training Doctoral Program. Her previous research interest was the self-renewal and differentiation of the neural stem cells. Now She is studying the mechanisms of direct lineage reprogramming of fibroblasts into neurons. Yingfei was great to have in the lab. COVID19 also affected Yingfei's carreer. In the midst of great progress COVID19 precluded much of her continuing to work. She went back to China to arrange for her PhD defense and we are hoping to be able to continue to work with her in the future. Good luck Yingfei!
I completed my PhD in Dr. TY Chang’s lab at Dartmouth College, where I studied cholesterol metabolism in health and disease. In the Wernig lab, my research focus is on studying neurological diseases in human neurons and other neural lineages. I am also interested in developing novel therapeutic approaches for treating incurable neurological disorders using reprogramming technology.
Yongjin received his Ph.D. in functional genomics in 2018 from the Seoul National University, South Korea. For his Ph.D., he focused on genetic mutations of human neurological patients and its functional impacts. Yongjin joined the Wernig lab in October 2018. In the Wernig lab, he is interested in developing therapeutic methods for neurological patients and understanding disease mechanisms using human stem cells and neurons. We are proud that Yongjin accepted an offer to join the faculty at the prestigious Seoul University.
Press
Wernig Lab Protocols & Recipes
Contact
We are always interested to hear from ambitious scientists and potential collaborators.
Marius Wernig
wernig@stanford.edu
Amy Lang, Lab Admin
alang2@stanford.edu
Institute for Stem Cell Biology and Regenerative Medicine
Wernig Laboratory
265 Campus Drive G3141
Stanford, CA 94305
U.S.A.
