

Wernig Laboratory

October 9, 2024: Welcome new postdoc Kirill to the lab!
Sep 17, 2024: Jinzhao's paper accepted in principle!
Jul 11, 2024: Gernot's paper published in Nature Communications!
March 21, 2024: Marius Mader's paper comes out in Nature Neuroscience!
Oct 10, 2023: Yongjin receives the Sammy Kuo Award from the Neuroscience Institute - CONGRATULATIONS!
Yongjin's paper on cell therapy in a mouse AD model is published in Cell Stem Cells
Our lab is generally interested in the molecular mechanisms that determine cell fates
Recently, we have identified a pool of transcription factors that are sufficient to convert skin fibroblasts directly into functional neuronal cells that we termed induced neuronal (iN) cells. This was a surprising finding and indicated that direct lineage reprogramming may be applicable to many somatic cell types and many different directions. Indeed, following our work others have identified transcription factors that could induce cardiomyocytes, blood progenitors, and hepatocytes from fibroblasts.
We are now focussing on two major aspects of iN and iPS cell reprogramming:
(i) we are fascinated by the puzzle how a hand full of transcription factors can so efficiently reprogram the entire epigenome of a cell so that it changes identity. To that end we are applying genome-wide expression analysis, chromatin immunoprecipitation, protein biochemistry, proteomics and functional screens.
(ii) it is equally exciting to now use reprogramming methods as tools to study or treat certain diseases. iPS cells have the great advantage that they can easily be genetically manipulated rendering them ideal for treating monogenetic disorders when combined with cell transplantation-based therapies. In particular we are working on Dystrophic Epidermolysis Bullosa in collaboration with Stanford's Dermatology Department. An exciting application of iN cell technology will be to try modeling neurological diseases in vitro. We perform both mouse and human experiments hoping to identify quantifiable phenotypes correlated with genotype and in a second step evaluate whether this assay could be used to discover novel drugs improve the disease progression.
Wernig Lab Research
Overview
Our lab is interested in the molecular mechanisms that define neural lineage identity focusing on transcription factors and chromatin biology. We use cellular reprogramming to understand how neurons are induced, how they mature and maintain their identity. Reprogramming also allows us to generate a novel tool box to study human neuronal and glial cell biology which become powerful human disease models in combination with genetic engineering. We further seek to develop reprogramming & genetic engineering approaches towards stem cell-based therapies. Finally, we study microglia-neuron interactions with the ultimate goal to understand the brain's immune system in health and disease and to exploit microglia for therapeutic and regenerative purposes.
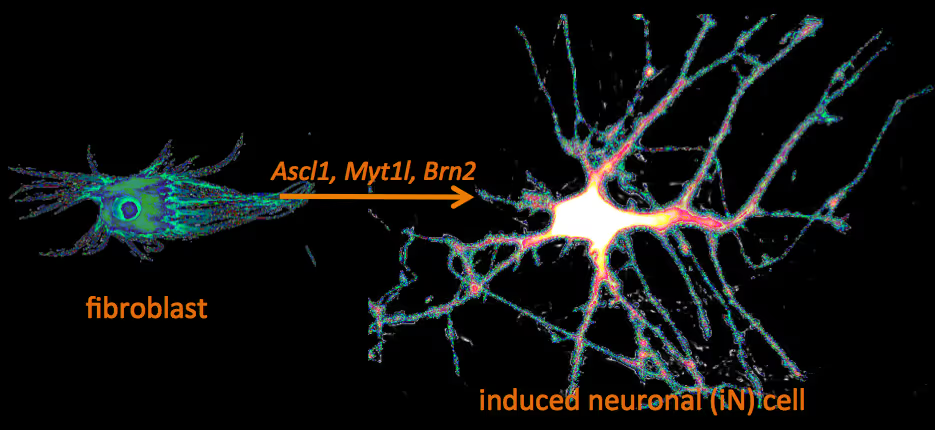
Human neuronal cell disease modeling
Neurosychiatric diseases like autism and schizophrenia are highly complex brain disorders difficult to model in mice in part due to complex genetic etiology and sometimes affecting human-specific genes. We develop novel human cell models to investigate disease-relevant cell biological phenomena.
Generation of defined human neuronal cell types to study neuronal cell biology
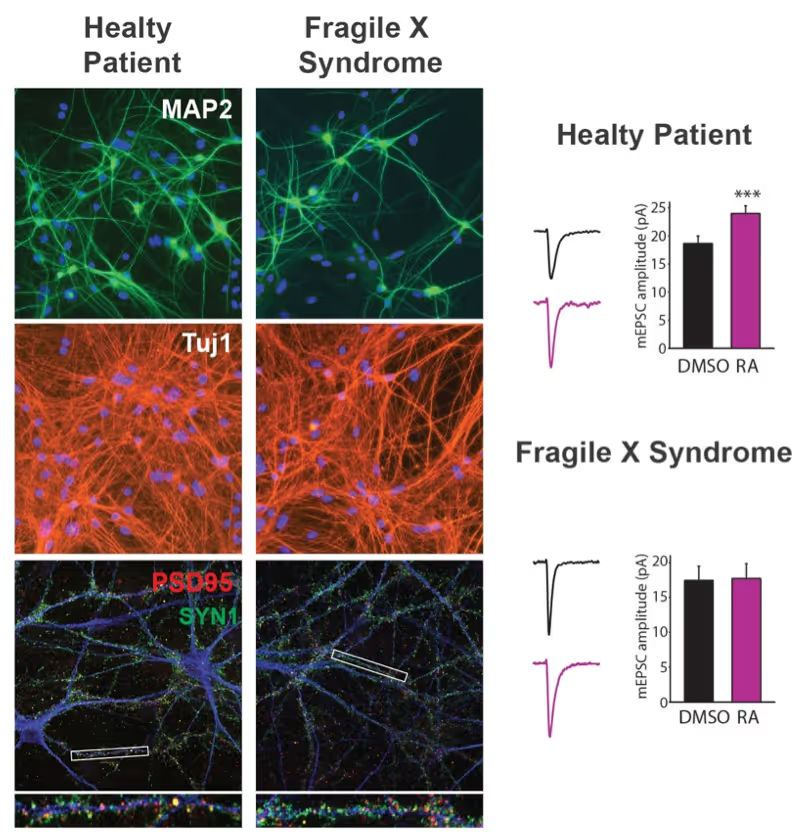
We have and continue to develop protocols to generate specific types of neurons such as pure glutamatergic and pure GABAergic neurons from human pluripotent stem cells using transcription factors. In combination with genetic engineering or deriving iPS cells from patients, we then interrogate the cell biology of human neurons that carry disease-causing mutations. A particular focus is on synaptic function as shown in the figure on the right on Fragile X Syndrome neurons in collaboration with Lu Chen and Tom Südhof's laboratories.
Making neurons from blood
The ability to generate functional induced neuronal cells from distantly related somatic cell types is fascinating but also offers the opportunity to obtain neurons from a larger cohort of human subjects. In particular blood is readily available and we showed can be efficiently converted into functional neurons from young and aged donors.
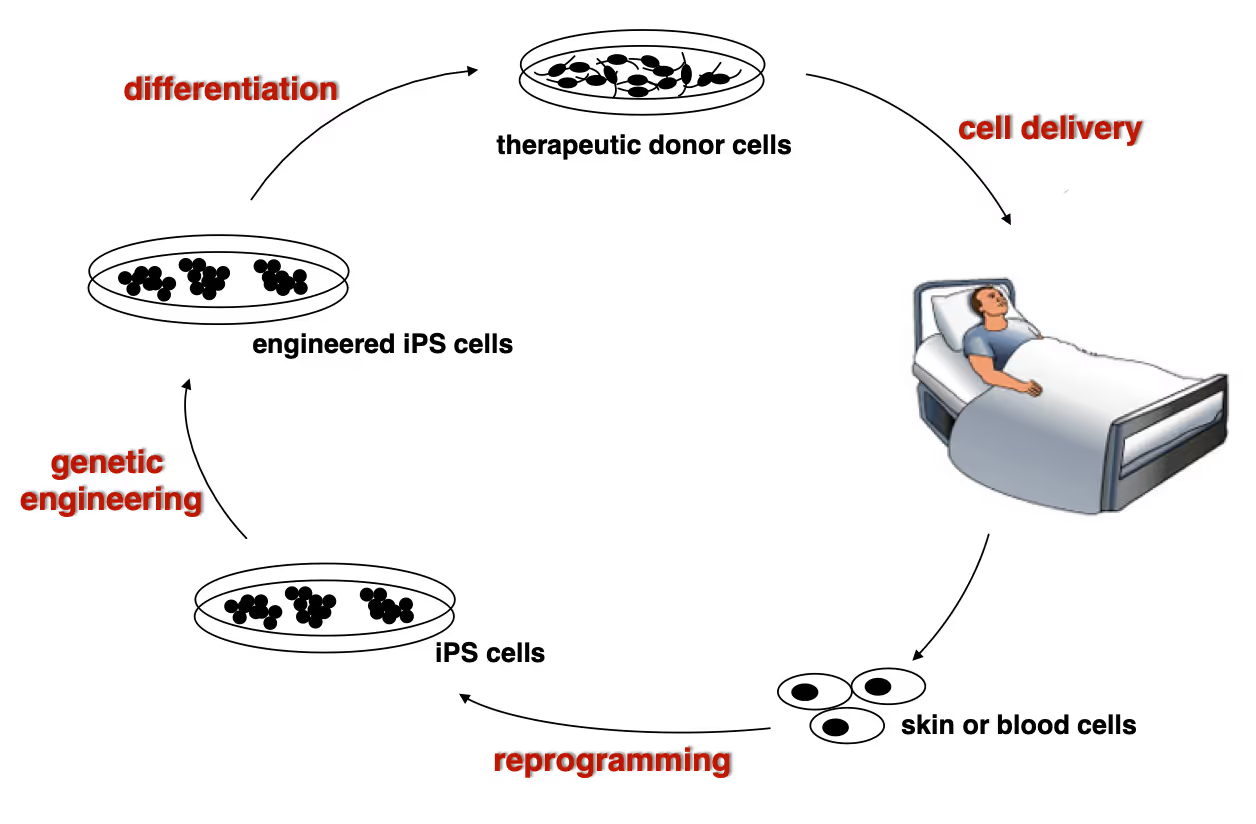
Developing next generation cell therapies
The combination of reprogramming and gene editing is truly powerful as it provides exciting new possibilities to generate cells that can be transplanted and have disease modifying activity. We currently apply this approach to restore mono-genetic diseases, but our vision goes beyond simple regenerative medicine. We will be able to genetically engineer designer cells that functionally integrate into diseased tissue equipped with sensing and intelligent disease-response mechanisms.
Towards a Phase 1 clinical trial for the fatal skin disease Epidermolysis Bullosa
Dystrophic Epidermolysis Bullosa is a severe, blistering monogenetic skin disease caused by mutations in the gene coding for type VII collagen. We have developed a 1-step gene editing/iPS cell reprogramming method to rapidly generate patient iPS cells corrected for their disease-causing mutations in the Collagen7a1 gene. In collaboration with dermatologist Tony Oro we are developing a cell manufacturing process compatible with Good Manufacturing Procedures (GMP) to obtain FDA-approval for a first in man Phase I clinical trial with with a genetically engineered iPS cell product.
Exploiting glia cell transplantation to treat neurodegenerative disease
Both oligodendrocyte precursor cells as well as microglia can efficiently repopulate the brain. We are interested in exploiting the properties of these cells to develop novel cell therapies for the brain either to use the transplanted cells to restore function such as myelination, to alter the function of transplanted cells for therepeutic benefit, to use the cells as vehicles for therapeutic molecules, or ultimately to develop designer cells that are engineered with genetic synthetic biology circuits to sense and interfere with disease processes of the brain.
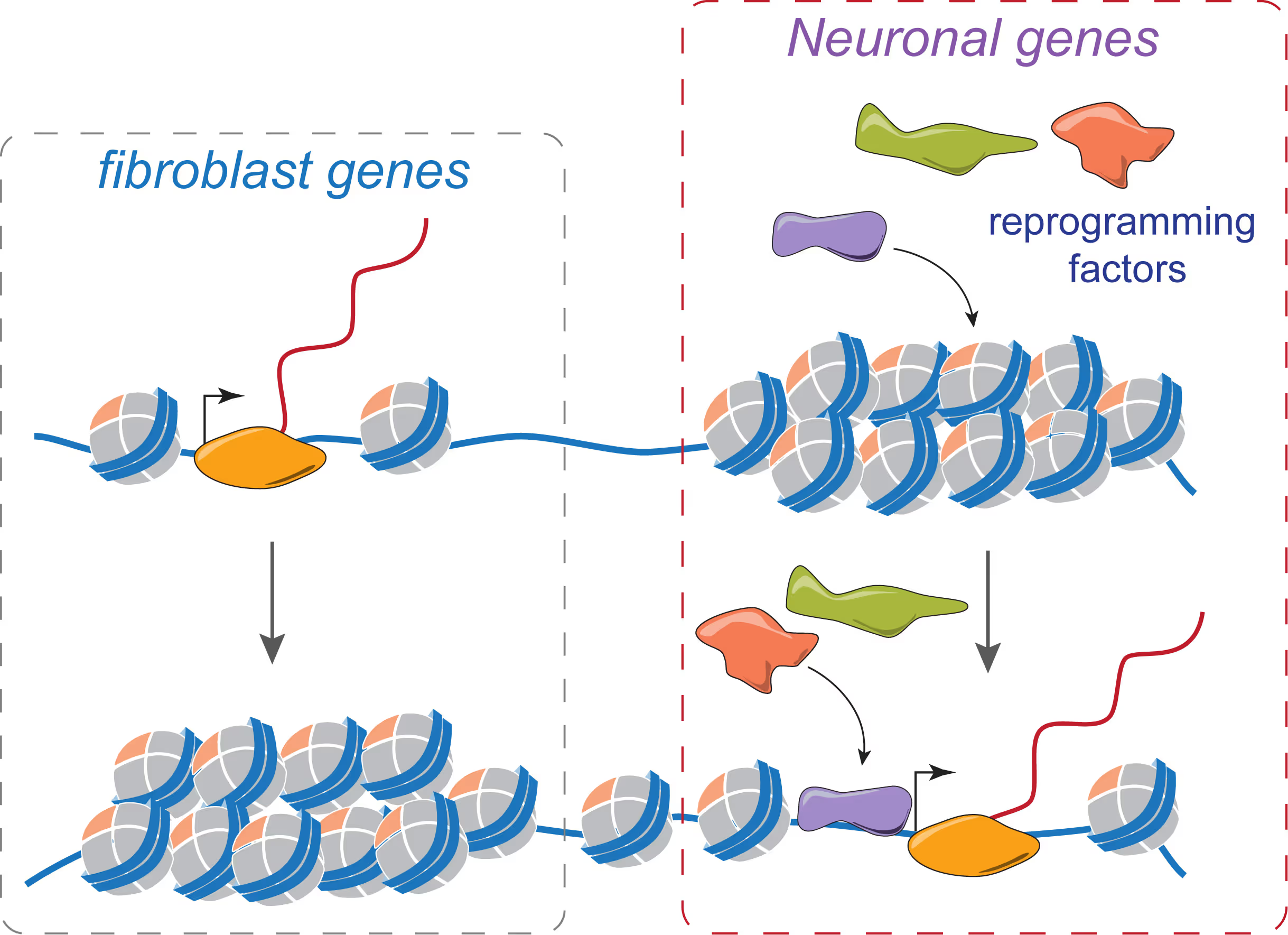
Mechanisms of neural cell lineage identity
We are interested in the molecular mechanisms that define neuronal and glial cell identity. We found sets of transcription factors that can convert fibroblasts or lymphocytes into neurons and oligodendrocytes. These factors are also operational during normal development and are largely responsible to induce terminal lineages from progenitor cells.
"On target" pioneer factors and chromatin remodeling during neuronal induction
We found that Ascl1, one of our reprogramming factors, has a unique ability to access its physiological targets even in fibroblasts where these sites are in a closed chromatin configuration. We are fascinated by this "on target" pioneering property and are investigating how Ascl1 can access its target sites in an unfavorable chromatin environment and how it then remodels the chromatin at these sites to activate the neuronal transcriptional program.
Maintenance of neuronal identity
Once neurons are made, there ought to be also mechanisms that maintain neuronal identity. We stumbled upon a novel repressive mechanism: The neuronal-specific transcription factor Myt1l continuosly represses many non-neuronal programs in neurons leaving the neuronal program open to activate by other factors and thereby ensuring stable neuronal gene expression. Myt1l was also recently found to be mutated in autism and schizophrenia.
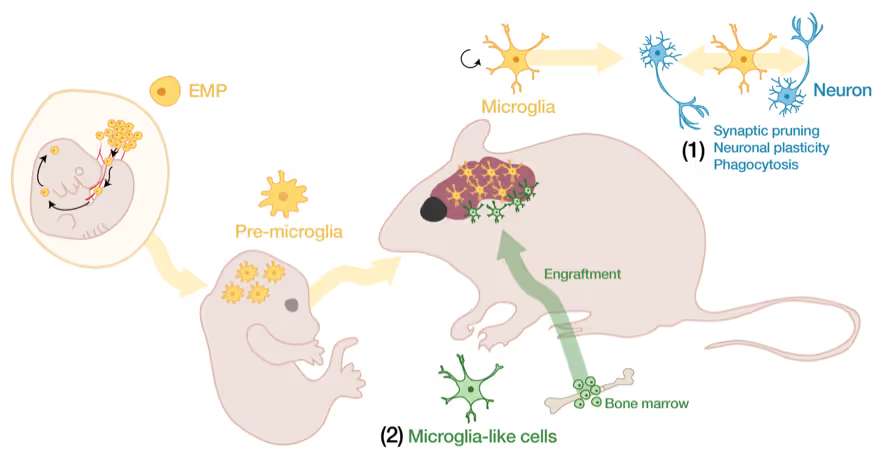
Microglia-neuron interactions in the healthy and diseased brain
Microglia, the brain's resident immune cells, are fascinating cells. They are derived from yolk sac progenitor cells early during development, are long-lived, and are not exchanged from bone marrow progenitor cells under physiological conditions. Microglia have been implicated in synaptic pruning, adult neurogenesis, and various brain diseases including Alzheimer's disease and Schizophrenia.
Developing an efficient microglia replacement system
We have developed a method to efficiently replace endogenous microglia from circulating cells without genetic manipulation. This does not happen physiologically but under certain conditions peripheral blood cells cross the blood-brain-barrier, migrate into the brain parenchyma and replace endogenous cells. We are investigating the cellular and molecular signals that enable circulating cells to invade the brain in order to further improve microglia replacement strategies.
The role of microglia in the normal and the diseased brain
Our ability to replace microglia provides us with a powerful tool to functionally perturb microglia function in normal and disease states. E.g. the microglial gene TREM2 is a strong Alzheimer's disease risk gene, but major questions about the neuro-immune interplay in the context of neurodegeneration and aging remain unsolved. Microglia replacement also provides an exciting prospect to develop novel cell therapies for a variety of brain diseases including enzyme deficiency syndromes, neurodegeneration, and brain tumors.
Lab Gene Expression Data
Publications
Zijian Zhang, Nicolas Denans, Yingfei Liu, Olena Zhulyn, Hannah D Rosenblatt, Marius Wernig, Maria Barna
Cells achieve highly efficient and accurate communication through cellular projections such as neurites and filopodia, yet there is a lack of genetically encoded tools that can selectively manipulate their composition and dynamics. Here, we present a versatile optogenetic toolbox of artificial multi-headed myosin motors that can move bidirectionally within long cellular extensions and allow for the selective transport of GFP-tagged cargo with light. Utilizing these engineered motors, we could transport bulky transmembrane receptors and organelles as well as actin remodellers to control the dynamics of both filopodia and neurites. Using an optimized in vivo imaging scheme, we further demonstrate that, upon limb amputation in axolotls, a complex array of filopodial extensions is formed. We selectively modulated these filopodial extensions and showed that they re-establish a Sonic Hedgehog signalling gradient during regeneration. Considering the ubiquitous existence of actin-based extensions, this toolbox shows the potential to manipulate cellular communication with unprecedented accuracy.
Cells achieve highly efficient and accurate communication through cellular projections such as neurites and filopodia, yet there is a lack of genetically encoded tools that can selectively manipulate their composition and dynamics. Here, we present a versatile optogenetic toolbox of artificial multi-headed myosin motors that can move bidirectionally within long cellular extensions and allow for the selective transport of GFP-tagged cargo with light. Utilizing these engineered motors, we could transport bulky transmembrane receptors and organelles as well as actin remodellers to control the dynamics of both filopodia and neurites. Using an optimized in vivo imaging scheme, we further demonstrate that, upon limb amputation in axolotls, a complex array of filopodial extensions is formed. We selectively modulated these filopodial extensions and showed that they re-establish a Sonic Hedgehog signalling gradient during regeneration. Considering the ubiquitous existence of actin-based extensions, this toolbox shows the potential to manipulate cellular communication with unprecedented accuracy.
Ng YH, Chanda S, Janas JA, Yang N, Kokubu Y, Südhof TC, Wernig M.
The differentiation of pluripotent stem cells can be accomplished by sequential activation of signaling pathways or through transcription factor programming. Multistep differentiation imitates embryonic development to obtain authentic cell types, but it suffers from asynchronous differentiation with variable efficiency. Transcription factor programming induces synchronous and efficient differentiation with higher reproducibility but may not always yield authentic cell types. We systematically explored the generation of dopaminergic induced neuronal cells from mouse and human pluripotent stem cells. We found that the proneural factor Ascl1 in combination with mesencephalic factors Lmx1a and Nurr1 induce peripheral dopaminergic neurons. Co-delivery of additional midbrain transcription factors En1, FoxA2, and Pitx3 resulted in facile and robust generation of functional dopaminergic neurons of midbrain character. Our results suggest that more complex combinations of transcription factors may be needed for proper regional specification of induced neuronal cells generated by direct lineage induction.
The differentiation of pluripotent stem cells can be accomplished by sequential activation of signaling pathways or through transcription factor programming. Multistep differentiation imitates embryonic development to obtain authentic cell types, but it suffers from asynchronous differentiation with variable efficiency. Transcription factor programming induces synchronous and efficient differentiation with higher reproducibility but may not always yield authentic cell types. We systematically explored the generation of dopaminergic induced neuronal cells from mouse and human pluripotent stem cells. We found that the proneural factor Ascl1 in combination with mesencephalic factors Lmx1a and Nurr1 induce peripheral dopaminergic neurons. Co-delivery of additional midbrain transcription factors En1, FoxA2, and Pitx3 resulted in facile and robust generation of functional dopaminergic neurons of midbrain character. Our results suggest that more complex combinations of transcription factors may be needed for proper regional specification of induced neuronal cells generated by direct lineage induction.
Pak C, Danko T, Mirabella VR, Wang J, Liu Y, Vangipuram M, Grieder S, Zhang X, Ward T, Huang YA, Jin K, Dexheimer P, Bardes E, Mitelpunkt A, Ma J, McLachlan M, Moore JC, Qu P, Purmann C, Dage JL, Swanson BJ, Urban AE, Aronow BJ, Pang ZP, Levinson DF, Wernig M, Südhof TC.
Heterozygous NRXN1 deletions constitute the most prevalent currently known single-gene mutation associated with schizophrenia, and additionally predispose to multiple other neurodevelopmental disorders. Engineered heterozygous NRXN1 deletions impaired neurotransmitter release in human neurons, suggesting a synaptic pathophysiological mechanism. Utilizing this observation for drug discovery, however, requires confidence in its robustness and validity. Here, we describe a multicenter effort to test the generality of this pivotal observation, using independent analyses at two laboratories of patient-derived and newly engineered human neurons with heterozygous NRXN1 deletions. Using neurons transdifferentiated from induced pluripotent stem cells that were derived from schizophrenia patients carrying heterozygous NRXN1 deletions, we observed the same synaptic impairment as in engineered NRXN1-deficient neurons. This impairment manifested as a large decrease in spontaneous synaptic events, in evoked synaptic responses, and in synaptic paired-pulse depression. Nrxn1-deficient mouse neurons generated from embryonic stem cells by the same method as human neurons did not exhibit impaired neurotransmitter release, suggesting a human-specific phenotype. Human NRXN1 deletions produced a reproducible increase in the levels of CASK, an intracellular NRXN1-binding protein, and were associated with characteristic gene-expression changes. Thus, heterozygous NRXN1 deletions robustly impair synaptic function in human neurons regardless of genetic background, enabling future drug discovery efforts.
Heterozygous NRXN1 deletions constitute the most prevalent currently known single-gene mutation associated with schizophrenia, and additionally predispose to multiple other neurodevelopmental disorders. Engineered heterozygous NRXN1 deletions impaired neurotransmitter release in human neurons, suggesting a synaptic pathophysiological mechanism. Utilizing this observation for drug discovery, however, requires confidence in its robustness and validity. Here, we describe a multicenter effort to test the generality of this pivotal observation, using independent analyses at two laboratories of patient-derived and newly engineered human neurons with heterozygous NRXN1 deletions. Using neurons transdifferentiated from induced pluripotent stem cells that were derived from schizophrenia patients carrying heterozygous NRXN1 deletions, we observed the same synaptic impairment as in engineered NRXN1-deficient neurons. This impairment manifested as a large decrease in spontaneous synaptic events, in evoked synaptic responses, and in synaptic paired-pulse depression. Nrxn1-deficient mouse neurons generated from embryonic stem cells by the same method as human neurons did not exhibit impaired neurotransmitter release, suggesting a human-specific phenotype. Human NRXN1 deletions produced a reproducible increase in the levels of CASK, an intracellular NRXN1-binding protein, and were associated with characteristic gene-expression changes. Thus, heterozygous NRXN1 deletions robustly impair synaptic function in human neurons regardless of genetic background, enabling future drug discovery efforts.
Raj N, McEachin ZT, Harousseau W, Zhou Y, Zhang F, Merritt-Garza ME, Taliaferro JM, Kalinowska M, Marro SG, Hales CM, Berry-Kravis E, Wolf-Ochoa MW, Martinez-Cerdeño V, Wernig M, Chen L, Klann E, Warren ST, Jin P, Wen Z, Bassell GJ.
Transcriptional silencing of the FMR1 gene in fragile X syndrome (FXS) leads to the loss of the RNA-binding protein FMRP. In addition to regulating mRNA translation and protein synthesis, emerging evidence suggests that FMRP acts to coordinate proliferation and differentiation during early neural development. However, whether loss of FMRP-mediated translational control is related to impaired cell fate specification in the developing human brain remains unknown. Here, we use human patient induced pluripotent stem cell (iPSC)-derived neural progenitor cells and organoids to model neurogenesis in FXS. We developed a high-throughput, in vitro assay that allows for the simultaneous quantification of protein synthesis and proliferation within defined neural subpopulations. We demonstrate that abnormal protein synthesis in FXS is coupled to altered cellular decisions to favor proliferative over neurogenic cell fates during early development. Furthermore, pharmacologic inhibition of elevated phosphoinositide 3-kinase (PI3K) signaling corrects both excess protein synthesis and cell proliferation in a subset of patient neural cells.
Transcriptional silencing of the FMR1 gene in fragile X syndrome (FXS) leads to the loss of the RNA-binding protein FMRP. In addition to regulating mRNA translation and protein synthesis, emerging evidence suggests that FMRP acts to coordinate proliferation and differentiation during early neural development. However, whether loss of FMRP-mediated translational control is related to impaired cell fate specification in the developing human brain remains unknown. Here, we use human patient induced pluripotent stem cell (iPSC)-derived neural progenitor cells and organoids to model neurogenesis in FXS. We developed a high-throughput, in vitro assay that allows for the simultaneous quantification of protein synthesis and proliferation within defined neural subpopulations. We demonstrate that abnormal protein synthesis in FXS is coupled to altered cellular decisions to favor proliferative over neurogenic cell fates during early development. Furthermore, pharmacologic inhibition of elevated phosphoinositide 3-kinase (PI3K) signaling corrects both excess protein synthesis and cell proliferation in a subset of patient neural cells.
Haag D, Mack N, Benites Goncalves da Silva P, Statz B, Clark J, Tanabe K, Sharma T, Jäger N, Jones DTW, Kawauchi D, Wernig M, Pfister SM
Diffuse intrinsic pontine glioma (DIPG) is an aggressive childhood tumor of the brainstem with currently no curative treatment available. The vast majority of DIPGs carry a histone H3 mutation leading to a lysine 27-to-methionine exchange (H3K27M). We engineered human induced pluripotent stem cells (iPSCs) to carry an inducible H3.3-K27M allele in the endogenous locus and studied the effects of the mutation in different disease-relevant neural cell types. H3.3-K27M upregulated bivalent promoter-associated developmental genes, producing diverse outcomes in different cell types. While being fatal for iPSCs, H3.3-K27M increased proliferation in neural stem cells (NSCs) and to a lesser extent in oligodendrocyte progenitor cells (OPCs). Only NSCs gave rise to tumors upon induction of H3.3-K27M and TP53 inactivation in an orthotopic xenograft model recapitulating human DIPGs. In NSCs, H3.3-K27M leads to maintained expression of stemness and proliferative genes and a premature activation of OPC programs that together may cause tumor initiation.
Diffuse intrinsic pontine glioma (DIPG) is an aggressive childhood tumor of the brainstem with currently no curative treatment available. The vast majority of DIPGs carry a histone H3 mutation leading to a lysine 27-to-methionine exchange (H3K27M). We engineered human induced pluripotent stem cells (iPSCs) to carry an inducible H3.3-K27M allele in the endogenous locus and studied the effects of the mutation in different disease-relevant neural cell types. H3.3-K27M upregulated bivalent promoter-associated developmental genes, producing diverse outcomes in different cell types. While being fatal for iPSCs, H3.3-K27M increased proliferation in neural stem cells (NSCs) and to a lesser extent in oligodendrocyte progenitor cells (OPCs). Only NSCs gave rise to tumors upon induction of H3.3-K27M and TP53 inactivation in an orthotopic xenograft model recapitulating human DIPGs. In NSCs, H3.3-K27M leads to maintained expression of stemness and proliferative genes and a premature activation of OPC programs that together may cause tumor initiation.
Saavedra L, Wallace K, Freudenrich T, Mall M, Mundy W, Davila J, Shafer T, Wernig M, Haag D
Assessment of neuroactive effects of chemicals in cell-based assays remains challenging as complex functional tissue is required for biologically relevant readouts. Recent in vitro models using rodent primary neural cultures grown on multielectrode arrays (MEAs) allow quantitative measurements of neural network activity suitable for neurotoxicity screening. However, robust systems for testing effects on network function in human neural models are still lacking. The increasing number of differentiation protocols for generating neurons from human induced pluripotent stem cells (hiPSCs) holds great potential to overcome the unavailability of human primary tissue and expedite cell-based assays. Yet, the variability in neuronal activity, prolonged ontogeny and rather immature stage of most neuronal cells derived by standard differentiation techniques greatly limit their utility for screening neurotoxic effects on human neural networks. Here, we used excitatory and inhibitory neurons, separately generated by direct reprogramming from hiPSCs, together with primary human astrocytes to establish highly functional cultures with defined cell ratios. Such neuron/glia co-cultures exhibited pronounced neuronal activity and robust formation of synchronized network activity on MEAs, albeit with noticeable delay compared to primary rat cortical cultures. We further investigated acute changes of network activity in human neuron/glia co-cultures and rat primary cortical cultures in response to compounds with known adverse neuroactive effects, including GABAA receptor antagonists and multiple pesticides. Importantly, we observed largely corresponding concentration-dependent effects on multiple neural network activity metrics using both neural culture types. These results demonstrate the utility of directly converted neuronal cells from hiPSCs for functional neurotoxicity screening of environmental chemicals.
Assessment of neuroactive effects of chemicals in cell-based assays remains challenging as complex functional tissue is required for biologically relevant readouts. Recent in vitro models using rodent primary neural cultures grown on multielectrode arrays (MEAs) allow quantitative measurements of neural network activity suitable for neurotoxicity screening. However, robust systems for testing effects on network function in human neural models are still lacking. The increasing number of differentiation protocols for generating neurons from human induced pluripotent stem cells (hiPSCs) holds great potential to overcome the unavailability of human primary tissue and expedite cell-based assays. Yet, the variability in neuronal activity, prolonged ontogeny and rather immature stage of most neuronal cells derived by standard differentiation techniques greatly limit their utility for screening neurotoxic effects on human neural networks. Here, we used excitatory and inhibitory neurons, separately generated by direct reprogramming from hiPSCs, together with primary human astrocytes to establish highly functional cultures with defined cell ratios. Such neuron/glia co-cultures exhibited pronounced neuronal activity and robust formation of synchronized network activity on MEAs, albeit with noticeable delay compared to primary rat cortical cultures. We further investigated acute changes of network activity in human neuron/glia co-cultures and rat primary cortical cultures in response to compounds with known adverse neuroactive effects, including GABAA receptor antagonists and multiple pesticides. Importantly, we observed largely corresponding concentration-dependent effects on multiple neural network activity metrics using both neural culture types. These results demonstrate the utility of directly converted neuronal cells from hiPSCs for functional neurotoxicity screening of environmental chemicals.
Michowski W, Chick JM, Chu C, Kolodziejczyk A, Wang Y, Suski JM, Abraham B, Anders L, Day D, Dunkl LM, Li Cheong Man M, Zhang T, Laphanuwat P, Bacon NA, Liu L, Fassl A, Sharma S, Otto T, Jecrois E, Han R, Sweeney KE, Marro S, Wernig M, Geng Y, Moses A, Li C, Gygi SP, Young RA, Sicinski P
The cyclin-dependent kinase 1 (Cdk1) drives cell division. To uncover additional functions of Cdk1, we generated knockin mice expressing an analog-sensitive version of Cdk1 in place of wild-type Cdk1. In our study, we focused on embryonic stem cells (ESCs), because this cell type displays particularly high Cdk1 activity. We found that in ESCs, a large fraction of Cdk1 substrates is localized on chromatin. Cdk1 phosphorylates many proteins involved in epigenetic regulation, including writers and erasers of all major histone marks. Consistent with these findings, inhibition of Cdk1 altered histone-modification status of ESCs. High levels of Cdk1 in ESCs phosphorylate and partially inactivate Dot1l, the H3K79 methyltransferase responsible for placing activating marks on gene bodies. Decrease of Cdk1 activity during ESC differentiation de-represses Dot1l, thereby allowing coordinated expression of differentiation genes. These analyses indicate that Cdk1 functions to maintain the epigenetic identity of ESCs.
The cyclin-dependent kinase 1 (Cdk1) drives cell division. To uncover additional functions of Cdk1, we generated knockin mice expressing an analog-sensitive version of Cdk1 in place of wild-type Cdk1. In our study, we focused on embryonic stem cells (ESCs), because this cell type displays particularly high Cdk1 activity. We found that in ESCs, a large fraction of Cdk1 substrates is localized on chromatin. Cdk1 phosphorylates many proteins involved in epigenetic regulation, including writers and erasers of all major histone marks. Consistent with these findings, inhibition of Cdk1 altered histone-modification status of ESCs. High levels of Cdk1 in ESCs phosphorylate and partially inactivate Dot1l, the H3K79 methyltransferase responsible for placing activating marks on gene bodies. Decrease of Cdk1 activity during ESC differentiation de-represses Dot1l, thereby allowing coordinated expression of differentiation genes. These analyses indicate that Cdk1 functions to maintain the epigenetic identity of ESCs.
Lee QY, Mall M, Chanda S, Zhou B, Sharma KS, Schaukowitch K, Adrian-Segarra JM, Grieder SD, Kareta MS, Wapinski OL, Ang CE, Li R, Südhof TC, Chang HY, Wernig M
The on-target pioneer factors Ascl1 and Myod1 are sequence-related but induce two developmentally unrelated lineages-that is, neuronal and muscle identities, respectively. It is unclear how these two basic helix-loop-helix (bHLH) factors mediate such fundamentally different outcomes. The chromatin binding of Ascl1 and Myod1 was surprisingly similar in fibroblasts, yet their transcriptional outputs were drastically different. We found that quantitative binding differences explained differential chromatin remodelling and gene activation. Although strong Ascl1 binding was exclusively associated with bHLH motifs, strong Myod1-binding sites were co-enriched with non-bHLH motifs, possibly explaining why Ascl1 is less context dependent. Finally, we observed that promiscuous binding of Myod1 to neuronal targets results in neuronal reprogramming when the muscle program is inhibited by Myt1l. Our findings suggest that chromatin access of on-target pioneer factors is primarily driven by the protein-DNA interaction, unlike ordinary context-dependent transcription factors, and that promiscuous transcription factor binding requires specific silencing mechanisms to ensure lineage fidelity.
The on-target pioneer factors Ascl1 and Myod1 are sequence-related but induce two developmentally unrelated lineages-that is, neuronal and muscle identities, respectively. It is unclear how these two basic helix-loop-helix (bHLH) factors mediate such fundamentally different outcomes. The chromatin binding of Ascl1 and Myod1 was surprisingly similar in fibroblasts, yet their transcriptional outputs were drastically different. We found that quantitative binding differences explained differential chromatin remodelling and gene activation. Although strong Ascl1 binding was exclusively associated with bHLH motifs, strong Myod1-binding sites were co-enriched with non-bHLH motifs, possibly explaining why Ascl1 is less context dependent. Finally, we observed that promiscuous binding of Myod1 to neuronal targets results in neuronal reprogramming when the muscle program is inhibited by Myt1l. Our findings suggest that chromatin access of on-target pioneer factors is primarily driven by the protein-DNA interaction, unlike ordinary context-dependent transcription factors, and that promiscuous transcription factor binding requires specific silencing mechanisms to ensure lineage fidelity.
Li L, Wang Y, Torkelson JL, Shankar G, Pattison JM, Zhen HH, Fang F, Duren Z, Xin J, Gaddam S, Melo SP, Piekos SN, Li J, Liaw EJ, Chen L, Li R, Wernig M, Wong WH, Chang HY, Oro AE
Tissue development results from lineage-specific transcription factors (TFs) programming a dynamic chromatin landscape through progressive cell fate transitions. Here, we define epigenomic landscape during epidermal differentiation of human pluripotent stem cells (PSCs) and create inference networks that integrate gene expression, chromatin accessibility, and TF binding to define regulatory mechanisms during keratinocyte specification. We found two critical chromatin networks during surface ectoderm initiation and keratinocyte maturation, which are driven by TFAP2C and p63, respectively. Consistently, TFAP2C, but not p63, is sufficient to initiate surface ectoderm differentiation, and TFAP2C-initiated progenitor cells are capable of maturing into functional keratinocytes. Mechanistically, TFAP2C primes the surface ectoderm chromatin landscape and induces p63 expression and binding sites, thus allowing maturation factor p63 to positively autoregulate its own expression and close a subset of the TFAP2C-initiated surface ectoderm program. Our work provides a general framework to infer TF networks controlling chromatin transitions that will facilitate future regenerative medicine advances.
Tissue development results from lineage-specific transcription factors (TFs) programming a dynamic chromatin landscape through progressive cell fate transitions. Here, we define epigenomic landscape during epidermal differentiation of human pluripotent stem cells (PSCs) and create inference networks that integrate gene expression, chromatin accessibility, and TF binding to define regulatory mechanisms during keratinocyte specification. We found two critical chromatin networks during surface ectoderm initiation and keratinocyte maturation, which are driven by TFAP2C and p63, respectively. Consistently, TFAP2C, but not p63, is sufficient to initiate surface ectoderm differentiation, and TFAP2C-initiated progenitor cells are capable of maturing into functional keratinocytes. Mechanistically, TFAP2C primes the surface ectoderm chromatin landscape and induces p63 expression and binding sites, thus allowing maturation factor p63 to positively autoregulate its own expression and close a subset of the TFAP2C-initiated surface ectoderm program. Our work provides a general framework to infer TF networks controlling chromatin transitions that will facilitate future regenerative medicine advances.
Ang CE, Ma Q, Wapinski OL, Fan S, Flynn RA, Lee QY, Coe B, Onoguchi M, Olmos VH, Do BT, Dukes-Rimsky L, Xu J, Tanabe K, Wang L, Elling U, Penninger JM, Zhao Y, Qu K, Eichler EE, Srivastava A, Wernig M, Chang HY.
Long noncoding RNAs (lncRNAs) have been shown to act as important cell biological regulators including cell fate decisions but are often ignored in human genetics. Combining differential lncRNA expression during neuronal lineage induction with copy number variation morbidity maps of a cohort of children with autism spectrum disorder/intellectual disability versus healthy controls revealed focal genomic mutations affecting several lncRNA candidate loci. Here we find that a t(5:12) chromosomal translocation in a family manifesting neurodevelopmental symptoms disrupts specifically lnc-NR2F1. We further show that lnc-NR2F1 is an evolutionarily conserved lncRNA functionally enhances induced neuronal cell maturation and directly occupies and regulates transcription of neuronal genes including autism-associated genes. Thus, integrating human genetics and functional testing in neuronal lineage induction is a promising approach for discovering candidate lncRNAs involved in neurodevelopmental diseases.
Long noncoding RNAs (lncRNAs) have been shown to act as important cell biological regulators including cell fate decisions but are often ignored in human genetics. Combining differential lncRNA expression during neuronal lineage induction with copy number variation morbidity maps of a cohort of children with autism spectrum disorder/intellectual disability versus healthy controls revealed focal genomic mutations affecting several lncRNA candidate loci. Here we find that a t(5:12) chromosomal translocation in a family manifesting neurodevelopmental symptoms disrupts specifically lnc-NR2F1. We further show that lnc-NR2F1 is an evolutionarily conserved lncRNA functionally enhances induced neuronal cell maturation and directly occupies and regulates transcription of neuronal genes including autism-associated genes. Thus, integrating human genetics and functional testing in neuronal lineage induction is a promising approach for discovering candidate lncRNAs involved in neurodevelopmental diseases.
Luo C, Lee QY, Wapinski O, Castanon R, Nery JR, Mall M, Kareta MS, Cullen SM, Goodell MA, Chang HY, Wernig M, Ecker JR.
Direct reprogramming of fibroblasts to neurons induces widespread cellular and transcriptional reconfiguration. Here, we characterized global epigenomic changes during the direct reprogramming of mouse fibroblasts to neurons using whole-genome base-resolution DNA methylation (mC) sequencing. We found that the pioneer transcription factor Ascl1 alone is sufficient for inducing the uniquely neuronal feature of non-CG methylation (mCH), but co-expression of Brn2 and Mytl1 was required to establish a global mCH pattern reminiscent of mature cortical neurons. Ascl1 alone induced promoter CG methylation (mCG) of fibroblast specific genes, while BAM overexpression additionally targets a competing myogenic program and directs a more faithful conversion to neuronal cells. Ascl1 induces local demethylation at its binding sites. Surprisingly, co-expression with Brn2 and Mytl1 inhibited the ability of Ascl1 to induce demethylation, suggesting a contextual regulation of transcription factor - epigenome interaction. Finally, we found that de novo methylation by DNMT3A is required for efficient neuronal reprogramming.
Direct reprogramming of fibroblasts to neurons induces widespread cellular and transcriptional reconfiguration. Here, we characterized global epigenomic changes during the direct reprogramming of mouse fibroblasts to neurons using whole-genome base-resolution DNA methylation (mC) sequencing. We found that the pioneer transcription factor Ascl1 alone is sufficient for inducing the uniquely neuronal feature of non-CG methylation (mCH), but co-expression of Brn2 and Mytl1 was required to establish a global mCH pattern reminiscent of mature cortical neurons. Ascl1 alone induced promoter CG methylation (mCG) of fibroblast specific genes, while BAM overexpression additionally targets a competing myogenic program and directs a more faithful conversion to neuronal cells. Ascl1 induces local demethylation at its binding sites. Surprisingly, co-expression with Brn2 and Mytl1 inhibited the ability of Ascl1 to induce demethylation, suggesting a contextual regulation of transcription factor - epigenome interaction. Finally, we found that de novo methylation by DNMT3A is required for efficient neuronal reprogramming.
Shariati SA, Dominguez A, Xie S, Wernig M, Qi LS, Skotheim JM.
The control of gene expression by transcription factor binding sites frequently determines phenotype. However, it is difficult to determine the function of single transcription factor binding sites within larger transcription networks. Here, we use deactivated Cas9 (dCas9) to disrupt binding to specific sites, a method we term CRISPRd. Since CRISPR guide RNAs are longer than transcription factor binding sites, flanking sequence can be used to target specific sites. Targeting dCas9 to an Oct4 site in the Nanog promoter displaced Oct4 from this site, reduced Nanog expression, and slowed division. In contrast, disrupting the Oct4 binding site adjacent to Pax6 upregulated Pax6 transcription and disrupting Nanog binding its own promoter upregulated its transcription. Thus, we can easily distinguish between activating and repressing binding sites and examine autoregulation. Finally, multiple guide RNA expression allows simultaneous inhibition of multiple binding sites, and conditionally destabilized dCas9 allows rapid reversibility.
The control of gene expression by transcription factor binding sites frequently determines phenotype. However, it is difficult to determine the function of single transcription factor binding sites within larger transcription networks. Here, we use deactivated Cas9 (dCas9) to disrupt binding to specific sites, a method we term CRISPRd. Since CRISPR guide RNAs are longer than transcription factor binding sites, flanking sequence can be used to target specific sites. Targeting dCas9 to an Oct4 site in the Nanog promoter displaced Oct4 from this site, reduced Nanog expression, and slowed division. In contrast, disrupting the Oct4 binding site adjacent to Pax6 upregulated Pax6 transcription and disrupting Nanog binding its own promoter upregulated its transcription. Thus, we can easily distinguish between activating and repressing binding sites and examine autoregulation. Finally, multiple guide RNA expression allows simultaneous inhibition of multiple binding sites, and conditionally destabilized dCas9 allows rapid reversibility.
Marro SG, Chanda S, Yang N, Janas JA, Valperga G, Trotter J, Zhou B, Merrill S, Yousif I, Shelby H, Vogel H, Kalani MYS, Südhof TC, Wernig M.
The autism-associated synaptic-adhesion gene Neuroligin-4 (NLGN4) is poorly conserved evolutionarily, limiting conclusions from Nlgn4 mouse models for human cells. Here, we show that the cellular and subcellular expression of human and murine Neuroligin-4 differ, with human Neuroligin-4 primarily expressed in cerebral cortex and localized to excitatory synapses. Overexpression of NLGN4 in human embryonic stem cell-derived neurons resulted in an increase in excitatory synapse numbers but a remarkable decrease in synaptic strength. Human neurons carrying the syndromic autism mutation NLGN4-R704C also formed more excitatory synapses but with increased functional synaptic transmission due to a postsynaptic mechanism, while genetic loss of NLGN4 did not significantly affect synapses in the human neurons analyzed. Thus, the NLGN4-R704C mutation represents a change-of-function mutation. Our work reveals contrasting roles of NLGN4 in human and mouse neurons, suggesting that human evolution has impacted even fundamental cell biological processes generally assumed to be highly conserved.
The autism-associated synaptic-adhesion gene Neuroligin-4 (NLGN4) is poorly conserved evolutionarily, limiting conclusions from Nlgn4 mouse models for human cells. Here, we show that the cellular and subcellular expression of human and murine Neuroligin-4 differ, with human Neuroligin-4 primarily expressed in cerebral cortex and localized to excitatory synapses. Overexpression of NLGN4 in human embryonic stem cell-derived neurons resulted in an increase in excitatory synapse numbers but a remarkable decrease in synaptic strength. Human neurons carrying the syndromic autism mutation NLGN4-R704C also formed more excitatory synapses but with increased functional synaptic transmission due to a postsynaptic mechanism, while genetic loss of NLGN4 did not significantly affect synapses in the human neurons analyzed. Thus, the NLGN4-R704C mutation represents a change-of-function mutation. Our work reveals contrasting roles of NLGN4 in human and mouse neurons, suggesting that human evolution has impacted even fundamental cell biological processes generally assumed to be highly conserved.
Nobuta H, Yang N, Ng YH, Marro SG, Sabeur K, Chavali M, Stockley JH, Killilea DW, Walter PB, Zhao C, Huie P Jr, Goldman SA, Kriegstein AR, Franklin RJM, Rowitch DH, Wernig M.
Pelizaeus-Merzbacher disease (PMD) is an X-linked leukodystrophy caused by mutations in Proteolipid Protein 1 (PLP1), encoding a major myelin protein, resulting in profound developmental delay and early lethality. Previous work showed involvement of unfolded protein response (UPR) and endoplasmic reticulum (ER) stress pathways, but poor PLP1 genotype-phenotype associations suggest additional pathogenetic mechanisms. Using induced pluripotent stem cell (iPSC) and gene-correction, we show that patient-derived oligodendrocytes can develop to the pre-myelinating stage, but subsequently undergo cell death. Mutant oligodendrocytes demonstrated key hallmarks of ferroptosis including lipid peroxidation, abnormal iron metabolism, and hypersensitivity to free iron. Iron chelation rescued mutant oligodendrocyte apoptosis, survival, and differentiationin vitro, and post-transplantation in vivo. Finally, systemic treatment of Plp1 mutant Jimpy mice with deferiprone, a small molecule iron chelator, reduced oligodendrocyte apoptosis and enabled myelin formation. Thus, oligodendrocyte iron-induced cell death and myelination is rescued by iron chelation in PMD pre-clinical models.
Pelizaeus-Merzbacher disease (PMD) is an X-linked leukodystrophy caused by mutations in Proteolipid Protein 1 (PLP1), encoding a major myelin protein, resulting in profound developmental delay and early lethality. Previous work showed involvement of unfolded protein response (UPR) and endoplasmic reticulum (ER) stress pathways, but poor PLP1 genotype-phenotype associations suggest additional pathogenetic mechanisms. Using induced pluripotent stem cell (iPSC) and gene-correction, we show that patient-derived oligodendrocytes can develop to the pre-myelinating stage, but subsequently undergo cell death. Mutant oligodendrocytes demonstrated key hallmarks of ferroptosis including lipid peroxidation, abnormal iron metabolism, and hypersensitivity to free iron. Iron chelation rescued mutant oligodendrocyte apoptosis, survival, and differentiationin vitro, and post-transplantation in vivo. Finally, systemic treatment of Plp1 mutant Jimpy mice with deferiprone, a small molecule iron chelator, reduced oligodendrocyte apoptosis and enabled myelin formation. Thus, oligodendrocyte iron-induced cell death and myelination is rescued by iron chelation in PMD pre-clinical models.
Essayan-Perez S, Zhou B, Nabet AM, Wernig M, Huang YA.
One in three people will develop Alzheimer's disease (AD) or another dementia and, despite intense research efforts, treatment options remain inadequate. Understanding the mechanisms of AD pathogenesis remains our principal hurdle to developing effective therapeutics to tackle this looming medical crisis. In light of recent discoveries from whole-genome sequencing and technical advances in humanized models, studying disease risk genes with induced human neural cells presents unprecedented advantages. Here, we first review the current knowledge of the proposed mechanisms underlying AD and focus on modern genetic insights to inform future studies. To highlight the utility of human pluripotent stem cell-based innovations, we then present an update on efforts in recapitulating the pathophysiology by induced neuronal, non-neuronal and a collection of brain cell types, departing from the neuron-centric convention. Lastly, we examine the translational potentials of such approaches, and provide our perspectives on the promise they offer to deepen our understanding of AD pathogenesis and to accelerate the development of intervention strategies for patients and risk carriers.
One in three people will develop Alzheimer's disease (AD) or another dementia and, despite intense research efforts, treatment options remain inadequate. Understanding the mechanisms of AD pathogenesis remains our principal hurdle to developing effective therapeutics to tackle this looming medical crisis. In light of recent discoveries from whole-genome sequencing and technical advances in humanized models, studying disease risk genes with induced human neural cells presents unprecedented advantages. Here, we first review the current knowledge of the proposed mechanisms underlying AD and focus on modern genetic insights to inform future studies. To highlight the utility of human pluripotent stem cell-based innovations, we then present an update on efforts in recapitulating the pathophysiology by induced neuronal, non-neuronal and a collection of brain cell types, departing from the neuron-centric convention. Lastly, we examine the translational potentials of such approaches, and provide our perspectives on the promise they offer to deepen our understanding of AD pathogenesis and to accelerate the development of intervention strategies for patients and risk carriers.
Ishii T, Ishikawa M, Fujimori K, Maeda T, Kushima I, Arioka Y, Mori D, Nakatake Y, Yamagata B, Nio S, Kato TA, Yang N, Wernig M, Kanba S, Mimura M, Ozaki N, Okano H.
Bipolar disorder (BP) and schizophrenia (SCZ) are major psychiatric disorders, but the molecular mechanisms underlying the complicated pathologies of these disorders remain unclear. It is difficult to establish adequate in vitro models for pathological analysis because of the heterogeneity of these disorders. In the present study, to recapitulate the pathologies of these disorders in vitro, we established in vitro models by differentiating mature neurons from human induced pluripotent stem cells (hiPSCs) derived from BP and SCZ patient with contributive copy number variations, as follows: two BP patients with PCDH15 deletion and one SCZ patient with RELN deletion. Glutamatergic neurons and GABAergic neurons were induced from hiPSCs under optimized conditions. Both types of induced neurons from both hiPSCs exhibited similar phenotypes of MAP2 (microtubule-associated protein 2)-positive dendrite shortening and decreasing synapse numbers. Additionally, we analyzed isogenic PCDH15- or RELN-deleted cells. The dendrite and synapse phenotypes of isogenic neurons were partially similar to those of patient-derived neurons. These results suggest that the observed phenotypes are general phenotypes of psychiatric disorders, and our in vitro models using hiPSC-based technology may be suitable for analysis of the pathologies of psychiatric disorders.
Bipolar disorder (BP) and schizophrenia (SCZ) are major psychiatric disorders, but the molecular mechanisms underlying the complicated pathologies of these disorders remain unclear. It is difficult to establish adequate in vitro models for pathological analysis because of the heterogeneity of these disorders. In the present study, to recapitulate the pathologies of these disorders in vitro, we established in vitro models by differentiating mature neurons from human induced pluripotent stem cells (hiPSCs) derived from BP and SCZ patient with contributive copy number variations, as follows: two BP patients with PCDH15 deletion and one SCZ patient with RELN deletion. Glutamatergic neurons and GABAergic neurons were induced from hiPSCs under optimized conditions. Both types of induced neurons from both hiPSCs exhibited similar phenotypes of MAP2 (microtubule-associated protein 2)-positive dendrite shortening and decreasing synapse numbers. Additionally, we analyzed isogenic PCDH15- or RELN-deleted cells. The dendrite and synapse phenotypes of isogenic neurons were partially similar to those of patient-derived neurons. These results suggest that the observed phenotypes are general phenotypes of psychiatric disorders, and our in vitro models using hiPSC-based technology may be suitable for analysis of the pathologies of psychiatric disorders.
Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, Srinivasan R, Li X, Devarajan K, Prélot L, Ang CE, Shibuya Y, Benayoun BA, Chang ALS, Wernig M, Wysocka J, Longaker MT, Snyder MP, Brunet A.
Age-associated chronic inflammation (inflammageing) is a central hallmark of ageing1, but its influence on specific cells remains largely unknown. Fibroblasts are present in most tissues and contribute to wound healing2,3. They are also the most widely used cell type for reprogramming to induced pluripotent stem (iPS) cells, a process that has implications for regenerative medicine and rejuvenation strategies4. Here we show that fibroblast cultures from old mice secrete inflammatory cytokines and exhibit increased variability in the efficiency of iPS cell reprogramming between mice. Variability between individuals is emerging as a feature of old age5-8, but the underlying mechanisms remain unknown. To identify drivers of this variability, we performed multi-omics profiling of fibroblast cultures from young and old mice that have different reprogramming efficiencies. This approach revealed that fibroblast cultures from old mice contain 'activated fibroblasts' that secrete inflammatory cytokines, and that the proportion of activated fibroblasts in a culture correlates with the reprogramming efficiency of that culture. Experiments in which conditioned medium was swapped between cultures showed that extrinsic factors secreted by activated fibroblasts underlie part of the variability between mice in reprogramming efficiency, and we have identified inflammatory cytokines, including TNF, as key contributors. Notably, old mice also exhibited variability in wound healing rate in vivo. Single-cell RNA-sequencing analysis identified distinct subpopulations of fibroblasts with different cytokine expression and signalling in the wounds of old mice with slow versus fast healing rates. Hence, a shift in fibroblast composition, and the ratio of inflammatory cytokines that they secrete, may drive the variability between mice in reprogramming in vitro and influence wound healing rate in vivo. This variability may reflect distinct stochastic ageing trajectories between individuals, and could help in developing personalized strategies to improve iPS cell generation and wound healing in elderly individuals.
Age-associated chronic inflammation (inflammageing) is a central hallmark of ageing1, but its influence on specific cells remains largely unknown. Fibroblasts are present in most tissues and contribute to wound healing2,3. They are also the most widely used cell type for reprogramming to induced pluripotent stem (iPS) cells, a process that has implications for regenerative medicine and rejuvenation strategies4. Here we show that fibroblast cultures from old mice secrete inflammatory cytokines and exhibit increased variability in the efficiency of iPS cell reprogramming between mice. Variability between individuals is emerging as a feature of old age5-8, but the underlying mechanisms remain unknown. To identify drivers of this variability, we performed multi-omics profiling of fibroblast cultures from young and old mice that have different reprogramming efficiencies. This approach revealed that fibroblast cultures from old mice contain 'activated fibroblasts' that secrete inflammatory cytokines, and that the proportion of activated fibroblasts in a culture correlates with the reprogramming efficiency of that culture. Experiments in which conditioned medium was swapped between cultures showed that extrinsic factors secreted by activated fibroblasts underlie part of the variability between mice in reprogramming efficiency, and we have identified inflammatory cytokines, including TNF, as key contributors. Notably, old mice also exhibited variability in wound healing rate in vivo. Single-cell RNA-sequencing analysis identified distinct subpopulations of fibroblasts with different cytokine expression and signalling in the wounds of old mice with slow versus fast healing rates. Hence, a shift in fibroblast composition, and the ratio of inflammatory cytokines that they secrete, may drive the variability between mice in reprogramming in vitro and influence wound healing rate in vivo. This variability may reflect distinct stochastic ageing trajectories between individuals, and could help in developing personalized strategies to improve iPS cell generation and wound healing in elderly individuals.
Huang YA, Zhou B, Nabet AM, Wernig M, Südhof TC.
In blood, apolipoprotein E (ApoE) is a component of circulating lipoproteins and mediates the clearance of these lipoproteins from blood by binding to ApoE receptors. Humans express three genetic ApoE variants, ApoE2, ApoE3, and ApoE4, which exhibit distinct ApoE receptor-binding properties and differentially affect Alzheimer's disease (AD), such that ApoE2 protects against, and ApoE4 predisposes to AD. In brain, ApoE-containing lipoproteins are secreted by activated astrocytes and microglia, but their functions and role in AD pathogenesis are largely unknown. Ample evidence suggests that ApoE4 induces microglial dysregulation and impedes Aβ clearance in AD, but the direct neuronal effects of ApoE variants are poorly studied. Extending previous studies, we here demonstrate that the three ApoE variants differentially activate multiple neuronal signaling pathways and regulate synaptogenesis. Specifically, using human neurons (male embryonic stem cell-derived) cultured in the absence of glia to exclude indirect glial mechanisms, we show that ApoE broadly stimulates signal transduction cascades. Among others, such stimulation enhances APP synthesis and synapse formation with an ApoE4>ApoE3>ApoE2 potency rank order, paralleling the relative risk for AD conferred by these ApoE variants. Unlike the previously described induction of APP transcription, however, ApoE-induced synaptogenesis involves CREB activation rather than cFos activation. We thus propose that in brain, ApoE acts as a glia-secreted signal that activates neuronal signaling pathways. The parallel potency rank order of ApoE4>ApoE3>ApoE2 in AD risk and neuronal signaling suggests that ApoE4 may in an apparent paradox promote AD pathogenesis by causing a chronic increase in signaling, possibly via enhancing APP expression.SIGNIFICANCE STATEMENT Humans express three genetic variants of apolipoprotein E (ApoE), ApoE2, ApoE3, and ApoE4. ApoE4 constitutes the most important genetic risk factor for Alzheimer's disease (AD), whereas ApoE2 protects against AD. Significant evidence suggests that ApoE4 impairs microglial function and impedes astrocytic Aβ clearance in brain, but the direct neuronal effects of ApoE are poorly understood, and the differences between ApoE variants in these effects are unclear. Here, we report that ApoE acts on neurons as a glia-secreted signaling molecule that, among others, enhances synapse formation. In activating neuronal signaling, the three ApoE variants exhibit a differential potency of ApoE4>ApoE3>ApoE2, which mirrors their relative effects on AD risk, suggesting that differential signaling by ApoE variants may contribute to AD pathogenesis.
In blood, apolipoprotein E (ApoE) is a component of circulating lipoproteins and mediates the clearance of these lipoproteins from blood by binding to ApoE receptors. Humans express three genetic ApoE variants, ApoE2, ApoE3, and ApoE4, which exhibit distinct ApoE receptor-binding properties and differentially affect Alzheimer's disease (AD), such that ApoE2 protects against, and ApoE4 predisposes to AD. In brain, ApoE-containing lipoproteins are secreted by activated astrocytes and microglia, but their functions and role in AD pathogenesis are largely unknown. Ample evidence suggests that ApoE4 induces microglial dysregulation and impedes Aβ clearance in AD, but the direct neuronal effects of ApoE variants are poorly studied. Extending previous studies, we here demonstrate that the three ApoE variants differentially activate multiple neuronal signaling pathways and regulate synaptogenesis. Specifically, using human neurons (male embryonic stem cell-derived) cultured in the absence of glia to exclude indirect glial mechanisms, we show that ApoE broadly stimulates signal transduction cascades. Among others, such stimulation enhances APP synthesis and synapse formation with an ApoE4>ApoE3>ApoE2 potency rank order, paralleling the relative risk for AD conferred by these ApoE variants. Unlike the previously described induction of APP transcription, however, ApoE-induced synaptogenesis involves CREB activation rather than cFos activation. We thus propose that in brain, ApoE acts as a glia-secreted signal that activates neuronal signaling pathways. The parallel potency rank order of ApoE4>ApoE3>ApoE2 in AD risk and neuronal signaling suggests that ApoE4 may in an apparent paradox promote AD pathogenesis by causing a chronic increase in signaling, possibly via enhancing APP expression.SIGNIFICANCE STATEMENT Humans express three genetic variants of apolipoprotein E (ApoE), ApoE2, ApoE3, and ApoE4. ApoE4 constitutes the most important genetic risk factor for Alzheimer's disease (AD), whereas ApoE2 protects against AD. Significant evidence suggests that ApoE4 impairs microglial function and impedes astrocytic Aβ clearance in brain, but the direct neuronal effects of ApoE are poorly understood, and the differences between ApoE variants in these effects are unclear. Here, we report that ApoE acts on neurons as a glia-secreted signaling molecule that, among others, enhances synapse formation. In activating neuronal signaling, the three ApoE variants exhibit a differential potency of ApoE4>ApoE3>ApoE2, which mirrors their relative effects on AD risk, suggesting that differential signaling by ApoE variants may contribute to AD pathogenesis.
Chanda S, Ang CE, Lee QY, Ghebrial M, Haag D, Shibuya Y, Wernig M, Südhof TC.
Human pluripotent stem cells can be rapidly converted into functional neurons by ectopic expression of proneural transcription factors. Here we show that directly reprogrammed neurons, despite their rapid maturation kinetics, can model teratogenic mechanisms that specifically affect early neurodevelopment. We delineated distinct phases of in vitro maturation during reprogramming of human neurons and assessed the cellular phenotypes of valproic acid (VPA), a teratogenic drug. VPA exposure caused chronic impairment of dendritic morphology and functional properties of developing neurons, but not those of mature neurons. These pathogenic effects were associated with VPA-mediated inhibition of the histone deacetylase (HDAC) and glycogen synthase kinase-3 (GSK-3) pathways, which caused transcriptional downregulation of many genes, including MARCKSL1, an actin-stabilizing protein essential for dendritic morphogenesis and synapse maturation during early neurodevelopment. Our findings identify a developmentally restricted pathogenic mechanism of VPA and establish the use of reprogrammed neurons as an effective platform for modeling teratogenic pathways.
Human pluripotent stem cells can be rapidly converted into functional neurons by ectopic expression of proneural transcription factors. Here we show that directly reprogrammed neurons, despite their rapid maturation kinetics, can model teratogenic mechanisms that specifically affect early neurodevelopment. We delineated distinct phases of in vitro maturation during reprogramming of human neurons and assessed the cellular phenotypes of valproic acid (VPA), a teratogenic drug. VPA exposure caused chronic impairment of dendritic morphology and functional properties of developing neurons, but not those of mature neurons. These pathogenic effects were associated with VPA-mediated inhibition of the histone deacetylase (HDAC) and glycogen synthase kinase-3 (GSK-3) pathways, which caused transcriptional downregulation of many genes, including MARCKSL1, an actin-stabilizing protein essential for dendritic morphogenesis and synapse maturation during early neurodevelopment. Our findings identify a developmentally restricted pathogenic mechanism of VPA and establish the use of reprogrammed neurons as an effective platform for modeling teratogenic pathways.
Tanabe K, Ang CE, Chanda S, Olmos VH, Haag D, Levinson DF, Südhof TC, Wernig M
Human cell models for disease based on induced pluripotent stem (iPS) cells have proven to be powerful new assets for investigating disease mechanisms. New insights have been obtained studying single mutations using isogenic controls generated by gene targeting. Modeling complex, multigenetic traits using patient-derived iPS cells is much more challenging due to line-to-line variability and technical limitations of scaling to dozens or more patients. Induced neuronal (iN) cells reprogrammed directly from dermal fibroblasts or urinary epithelia could be obtained from many donors, but such donor cells are heterogeneous, show interindividual variability, and must be extensively expanded, which can introduce random mutations. Moreover, derivation of dermal fibroblasts requires invasive biopsies. Here we show that human adult peripheral blood mononuclear cells, as well as defined purified T lymphocytes, can be directly converted into fully functional iN cells, demonstrating that terminally differentiated human cells can be efficiently transdifferentiated into a distantly related lineage. T cell-derived iN cells, generated by nonintegrating gene delivery, showed stereotypical neuronal morphologies and expressed multiple pan-neuronal markers, fired action potentials, and were able to form functional synapses. These cells were stable in the absence of exogenous reprogramming factors. Small molecule addition and optimized culture systems have yielded conversion efficiencies of up to 6.2%, resulting in the generation of >50,000 iN cells from 1 mL of peripheral blood in a single step without the need for initial expansion. Thus, our method allows the generation of sufficient neurons for experimental interrogation from a defined, homogeneous, and readily accessible donor cell population.
Human cell models for disease based on induced pluripotent stem (iPS) cells have proven to be powerful new assets for investigating disease mechanisms. New insights have been obtained studying single mutations using isogenic controls generated by gene targeting. Modeling complex, multigenetic traits using patient-derived iPS cells is much more challenging due to line-to-line variability and technical limitations of scaling to dozens or more patients. Induced neuronal (iN) cells reprogrammed directly from dermal fibroblasts or urinary epithelia could be obtained from many donors, but such donor cells are heterogeneous, show interindividual variability, and must be extensively expanded, which can introduce random mutations. Moreover, derivation of dermal fibroblasts requires invasive biopsies. Here we show that human adult peripheral blood mononuclear cells, as well as defined purified T lymphocytes, can be directly converted into fully functional iN cells, demonstrating that terminally differentiated human cells can be efficiently transdifferentiated into a distantly related lineage. T cell-derived iN cells, generated by nonintegrating gene delivery, showed stereotypical neuronal morphologies and expressed multiple pan-neuronal markers, fired action potentials, and were able to form functional synapses. These cells were stable in the absence of exogenous reprogramming factors. Small molecule addition and optimized culture systems have yielded conversion efficiencies of up to 6.2%, resulting in the generation of >50,000 iN cells from 1 mL of peripheral blood in a single step without the need for initial expansion. Thus, our method allows the generation of sufficient neurons for experimental interrogation from a defined, homogeneous, and readily accessible donor cell population.
Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Südhof TC, Wernig M, Chen L
Fragile X syndrome (FXS) is an X chromosome-linked disease leading to severe intellectual disabilities. FXS is caused by inactivation of the fragile X mental retardation 1 (FMR1) gene, but how FMR1 inactivation induces FXS remains unclear. Using human neurons generated from control and FXS patient-derived induced pluripotent stem (iPS) cells or from embryonic stem cells carrying conditional FMR1 mutations, we show here that loss of FMR1 function specifically abolished homeostatic synaptic plasticity without affecting basal synaptic transmission. We demonstrated that, in human neurons, homeostatic plasticity induced by synaptic silencing was mediated by retinoic acid, which regulated both excitatory and inhibitory synaptic strength. FMR1 inactivation impaired homeostatic plasticity by blocking retinoic acid-mediated regulation of synaptic strength. Repairing the genetic mutation in the FMR1 gene in an FXS patient cell line restored fragile X mental retardation protein (FMRP) expression and fully rescued synaptic retinoic acid signaling. Thus, our study reveals a robust functional impairment caused by FMR1 mutations that might contribute to neuronal dysfunction in FXS. In addition, our results suggest that FXS patient iPS cell-derived neurons might be useful for studying the mechanisms mediating functional abnormalities in FXS.
Fragile X syndrome (FXS) is an X chromosome-linked disease leading to severe intellectual disabilities. FXS is caused by inactivation of the fragile X mental retardation 1 (FMR1) gene, but how FMR1 inactivation induces FXS remains unclear. Using human neurons generated from control and FXS patient-derived induced pluripotent stem (iPS) cells or from embryonic stem cells carrying conditional FMR1 mutations, we show here that loss of FMR1 function specifically abolished homeostatic synaptic plasticity without affecting basal synaptic transmission. We demonstrated that, in human neurons, homeostatic plasticity induced by synaptic silencing was mediated by retinoic acid, which regulated both excitatory and inhibitory synaptic strength. FMR1 inactivation impaired homeostatic plasticity by blocking retinoic acid-mediated regulation of synaptic strength. Repairing the genetic mutation in the FMR1 gene in an FXS patient cell line restored fragile X mental retardation protein (FMRP) expression and fully rescued synaptic retinoic acid signaling. Thus, our study reveals a robust functional impairment caused by FMR1 mutations that might contribute to neuronal dysfunction in FXS. In addition, our results suggest that FXS patient iPS cell-derived neurons might be useful for studying the mechanisms mediating functional abnormalities in FXS.
Mesentier-Louro LA, Dodd R, Domizi P, Nobuta H, Wernig M, Wernig G, Liao YJ
BACKGROUND: Animal models of optic nerve injury are often used to study central nervous system (CNS) degeneration and regeneration, and targeting the optic nerve is a powerful approach for axon-protective or remyelination therapy. However, the experimental delivery of drugs or cells to the optic nerve is rarely performed because injections into this structure are difficult in small animals, especially in mice. NEW METHOD: We investigated and developed methods to deliver drugs or cells to the mouse optic nerve through 3 different routes: a) intraorbital, b) through the optic foramen and c) transcranial. RESULTS: The methods targeted different parts of the mouse optic nerve: intraorbital proximal (intraorbital), intracranial middle (optic-foramen) or intracranial distal (transcranial) portion. COMPARISON WITH EXISTING METHODS: Most existing methods target the optic nerve indirectly. For instance, intravitreally delivered cells often cannot cross the inner limiting membrane to reach retinal neurons and optic nerve axons. Systemic delivery, eye drops and intraventricular injections do not always successfully target the optic nerve. Intraorbital and transcranial injections into the optic nerve or chiasm have been performed but these methods have not been well described. We approached the optic nerve with more selective and precise targeting than existing methods. CONCLUSIONS: We successfully targeted the murine optic nerve intraorbitally, through the optic foramen, and transcranially. Of all methods, the injection through the optic foramen is likely the most innovative and fastest. These methods offer additional approaches for therapeutic intervention to be used by those studying white matter damage and axonal regeneration in the CNS.
BACKGROUND: Animal models of optic nerve injury are often used to study central nervous system (CNS) degeneration and regeneration, and targeting the optic nerve is a powerful approach for axon-protective or remyelination therapy. However, the experimental delivery of drugs or cells to the optic nerve is rarely performed because injections into this structure are difficult in small animals, especially in mice. NEW METHOD: We investigated and developed methods to deliver drugs or cells to the mouse optic nerve through 3 different routes: a) intraorbital, b) through the optic foramen and c) transcranial. RESULTS: The methods targeted different parts of the mouse optic nerve: intraorbital proximal (intraorbital), intracranial middle (optic-foramen) or intracranial distal (transcranial) portion. COMPARISON WITH EXISTING METHODS: Most existing methods target the optic nerve indirectly. For instance, intravitreally delivered cells often cannot cross the inner limiting membrane to reach retinal neurons and optic nerve axons. Systemic delivery, eye drops and intraventricular injections do not always successfully target the optic nerve. Intraorbital and transcranial injections into the optic nerve or chiasm have been performed but these methods have not been well described. We approached the optic nerve with more selective and precise targeting than existing methods. CONCLUSIONS: We successfully targeted the murine optic nerve intraorbitally, through the optic foramen, and transcranially. Of all methods, the injection through the optic foramen is likely the most innovative and fastest. These methods offer additional approaches for therapeutic intervention to be used by those studying white matter damage and axonal regeneration in the CNS.
Liu Y, Yu C, Daley TP, Wang F, Cao WS, Bhate S, Lin X, Still C, Liu H, Zhao D, Wang H, Xie XS, Ding S, Wong WH, Wernig M, Qi LS
Human cell models for disease based on induced pluripotent stem (iPS) cells have proven to be powerful new assets for investigating disease mechanisms. New insights have been obtained studying single mutations using isogenic controls generated by gene targeting. Modeling complex, multigenetic traits using patient-derived iPS cells is much more challenging due to line-to-line variability and technical limitations of scaling to dozens or more patients. Induced neuronal (iN) cells reprogrammed directly from dermal fibroblasts or urinary epithelia could be obtained from many donors, but such donor cells are heterogeneous, show interindividual variability, and must be extensively expanded, which can introduce random mutations. Moreover, derivation of dermal fibroblasts requires invasive biopsies. Here we show that human adult peripheral blood mononuclear cells, as well as defined purified T lymphocytes, can be directly converted into fully functional iN cells, demonstrating that terminally differentiated human cells can be efficiently transdifferentiated into a distantly related lineage. T cell-derived iN cells, generated by nonintegrating gene delivery, showed stereotypical neuronal morphologies and expressed multiple pan-neuronal markers, fired action potentials, and were able to form functional synapses. These cells were stable in the absence of exogenous reprogramming factors. Small molecule addition and optimized culture systems have yielded conversion efficiencies of up to 6.2%, resulting in the generation of >50,000 iN cells from 1 mL of peripheral blood in a single step without the need for initial expansion. Thus, our method allows the generation of sufficient neurons for experimental interrogation from a defined, homogeneous, and readily accessible donor cell population.
Human cell models for disease based on induced pluripotent stem (iPS) cells have proven to be powerful new assets for investigating disease mechanisms. New insights have been obtained studying single mutations using isogenic controls generated by gene targeting. Modeling complex, multigenetic traits using patient-derived iPS cells is much more challenging due to line-to-line variability and technical limitations of scaling to dozens or more patients. Induced neuronal (iN) cells reprogrammed directly from dermal fibroblasts or urinary epithelia could be obtained from many donors, but such donor cells are heterogeneous, show interindividual variability, and must be extensively expanded, which can introduce random mutations. Moreover, derivation of dermal fibroblasts requires invasive biopsies. Here we show that human adult peripheral blood mononuclear cells, as well as defined purified T lymphocytes, can be directly converted into fully functional iN cells, demonstrating that terminally differentiated human cells can be efficiently transdifferentiated into a distantly related lineage. T cell-derived iN cells, generated by nonintegrating gene delivery, showed stereotypical neuronal morphologies and expressed multiple pan-neuronal markers, fired action potentials, and were able to form functional synapses. These cells were stable in the absence of exogenous reprogramming factors. Small molecule addition and optimized culture systems have yielded conversion efficiencies of up to 6.2%, resulting in the generation of >50,000 iN cells from 1 mL of peripheral blood in a single step without the need for initial expansion. Thus, our method allows the generation of sufficient neurons for experimental interrogation from a defined, homogeneous, and readily accessible donor cell population.
Marius Wernig
wernig@stanford.edu
Dr. Marius Wernig is a Professor of Pathology and a Co-Director of the Institute for Stem Cell Biology and Regenerative Medicine at Stanford University. He graduated with an M.D. Ph.D. from the Technical University of Munich where he trained in developmental genetics in the lab of Rudi Balling. After completing his residency in Neuropathology and General Pathology at the University of Bonn, he then became a postdoctoral fellow in the lab of Dr. Rudolf Jaenisch at the Whitehead Institute for Biomedical Research/ MIT in Cambridge, MA. In 2008, Dr. Wernig joined the faculty of the Institute for Stem Cell Biology and Regenerative Medicine at Stanford University where he has been ever since.
He received an NIH Pathway to Independence Award, the Cozzarelli Prize for Outstanding Scientific Excellence from the National Academy of Sciences U.S.A., the Outstanding Investigator Award from the International Society for Stem Cell Research, the New York Stem Cell Foundation Robertson Stem Cell Prize, the Ogawa-Yamanaka Stem Cell Prize delivered by the Gladstone Institute and more recently has been named a Faculty Scholar by the Howard Hughes Medical Institute.
Dr. Wernig’s lab is interested in pluripotent stem cell biology and the molecular determinants of neural cell fate decisions. His laboratory was the first to generate functional neuronal cells reprogrammed directly from skin fibroblasts, which he termed induced neuronal (iN) cells. The lab is now working on identifying the molecular mechanisms underlying induced lineage fate changes, the phenotypic consequences of disease-causing mutations in human neurons and other neural lineages as well as the development of novel therapeutic gene targeting and cell transplantation-based strategies for a variety of monogenetic diseases.
Academic appointments
Associate Professor Institute for Stem Cell Biology and Regenerative Medicine
Member:
Bio-X
Cardiovascular Institute
Child Health Research Institute
Institute for Stem Cell Biology and Regenerative Medicine
Stanford Cancer Institute
Stanford Neurosciences Institute
Administrative appointments
Faculty Senate, Department of Pathology (2017 - Present)
Assistant Professor, Institute for Stem Cell Biology and Regenerative Medicine (2008 - 2014)
Honors & Awards
HHMI Faculty Scholar Award, Howard Hughes Medical Institute (2016)
New York Stem Cell Foundation Robertson Stem Cell Prize, New York Stem Cell Foundation (2014)
The Outstanding Young Investigator Award, International Society for Stem Cell Research (2013)
Ascina Award, Republic of Austria (2010)
Cozzarelli Prize for Outstanding Scientific Excellence, National Academy of Sciences USA (2009)
New Scholar in Aging, Ellison Medical Foundation (2010)
Robertson Investigator Award, New York Stem Cell Foundation (2010)
Donald E. and Delia B. Baxter Faculty Scholarship, Stanford University (2009)
Margaret and Herman Sokol Award, Biomedical Research (2007)
Longterm fellowship Human Frontiers Science Program Organisation, HFSP (2004-2006)
Boards, Advisory Committees
Professional Organizations Member, Society for Neuroscience (2003 - Present)
Member, International Society for Stem Cell Research (2004 - Present)
Editorial Board Member, Cell Stem Cell (2012 - Present)
Editorial Board Member, Stem Cell Reports (2013 - Present)
Member, Program Committee, Society for Neuroscience (2016 - Present)
Chair, Program Committee, International Society for Stem Cell Research (2017 - Present)
Professional Education
M.D., Technical University of Munich, Medicine (2000)
Team

I joined the Wernig lab as an LSRP in the summer of 2022 after graduating from UCLA. I’m interested in the regenerative capabilities of the human body and the molecular mechanisms underlying these regenerative systems, as well as the ways in which we can utilize these mechanisms to treat disease via tools such as cell and gene therapy. My research interests further include immunological topics such as cancer biology and the coevolution of pathogenic species and the immune response. In the Wernig lab, I am investigating methods of replacing dysfunctional microglia in the CNS in the context of neurodegenerative diseases, as well as the translational potential of gene editing and the role that genetically manipulated cells can play in treating such diseases. Outside the lab, I enjoy spending time at the beach, watching sports, and going on road trips with my friends.
Danwei Wu is a Stanford neurology resident in the Neuroscience Scholar Track and aspiring neuroimmunologist. Prior to starting residency, she completed an HHMI Medical Research Fellowship with Dr. Vann Bennett at Duke University studying neuron-specific membrane domains and their interaction with cytoskeletal structures. Her current research interest includes cell-based therapies for multiple sclerosis, molecular pathways of neuro-repair, and pathogenesis of autoimmunity. She is interested in developing new therapies for neurologic diseases. Outside of the lab, she enjoys hiking and reading science fiction.
I am a PhD student in the Neurosciences Interdepartmental Program and joined the lab in summer 2022. I am interested in developing methods to reprogram stem cells directly into oligodendrocytes. I then hope to use these cells to study the role of oligodendrocytes in aging and neurodegeneration. In my free time I like to read, ski, and play tennis.
I am a passionate cellular and molecular biologistwith expertise in research related to cancer, genomic/chromosomal instability, DNA damage response, epigenetics, proteomics and cellular identity. The latter topic attracted me to the Wernig lab where I aim to decipher the mechanisms that allow us to specifically switch the identity of a cell, converting differentiated somatic cells into induced neurons. I also work on a project that aims to integrate CRISPR/CAS9-mediated gene correction with iPS cell generation in order to establish a therapy for the devastating skin disease epidermolysis bullosa. In my free time I play underwater rugby, surf, spearfish and ski!
I am an undergraduate studying neurobiology and computer science. In the Wernig Lab, I am studying how the interactions between reprogramming factors and chromatin modifiers allow for fully developed fibrojacklynl@stanford.edublasts to reprogram into induced neuronal cells (iN).
I studied small GTPase signaling and how its perturbation can contribute to cancer and neurological disorders during my PhD training at Cold Spring Harbor Laboratory. Since then, I have become increasingly fascinated by the epigenetic mechanisms of gene regulation, and how changes in epigenome influence cell function and fate decisions. In the Wernig lab, I am currently investigating the interactions between the reprogramming factors and chromatin modifiers. More specifically, I am interested in finding out how such interactions enable a terminally differentiated cell—for example, a fibroblast—to acquire new transcriptional program that allows its reprogramming into induced neuron (iN).
I am a Bachelor of Science candidate in Bioengineering at Stanford University. My research in the Wernig Lag involves investigating the relationship between microglia and the IKBKAP gene, specifically its role in mechanisms such as microglia regeneration and the transdifferentiation of hematopoietic stem and progenitor cells into microglia-like cells.
Kayla is a PhD student in the Neurosciences Interdepartmental Program and joined the lab in summer 2022. She is interested in leveraging the lab's cell reprogramming techniques to study the roles of individual cell types in proteinopathies. She aims to faithfully recapitulate disease states and uncover the underlying molecular mechanisms that lead to neurodegeneration. In her free time, Kayla can be found birding, hiking, or performing with the Stanford Symphony Orchestra.
I received my BA in molecular and cell biology (Neurobiology focus) and computer science from UC Berkeley. I went on to do my PhD at Duke University, where I worked on engineering electrical synapses for precise neuromodulation in Kafui Dzirasa’s lab. Now, as a postdoc in Marius Wernig lab, I am interested in delving deeper into cellular engineering to better understand neuronal induction, synapse formation, and deliver treatment across the blood brain barrier using engineered microglia. In my free time, I enjoy cooking and coding/electrical engineering projects.
As a neurosurgical resident, my research interests include translational topics such as cell therapy and neuroregenerative mechanisms. In the Wernig lab, I study the cellular reprogramming potential of microglia. Moreover, I’m interested in the functional integration of induced neurons into neural systems.
I am an undergraduate student studying Human Biology. At Wernig Lab, I am helping to research how microglial cells organize and communicate with each other. I am interested in understanding the relationship between changes in the organization of microglia and other neural cells and the progression of neuronal diseases.
I received my Ph.D. in Neuroscience and Brain Technologies in 2021 from the University of Genoa and the Italian Institute of Technology, Italy. I joined the Wernig lab in December 2022. Here, I am interested in understanding how the microglia-derived factors end up in the neuronal lysosomes. Furthermore, I am interested in understanding how the accumulation and breakdown of these factors lead to neurodegenerative disorders using human induced pluripotent stem cell culture. I read non-fiction novels, sketch, and go for hikes in my free time.
Rida Rehman received her doctorate in Natural Sciences (Dr. Rer. Nat.), specializing in brain trauma and rehabilitation at Universtät Ulm, Germany in 2023. She also holds an M.S. in Biomedical Sciences and a B.S. in Applied Biosciences from NUST, Pakistan. Rida’s research has primarily focused on understanding the critical role of microglia in neurotrauma, degeneration, reprogramming and repair. Rida joined Wernig lab in 2023 and is primarily working on microglia to neuron reprogramming.
I worked as a neurologist for 10 years in Japan. During clinical practice, I saw a lot of patients who could not be treated by the current medicine. As a result, I became strongly attracted to basic research of neuroscience. I received my PhD in Dr. Tatsushi Toda’s lab at Kobe University, where I studied disease-modifying drug for Parkinson’s disease. As a post doc fellow in the Wernig lab, I’m interested in investigating the ubiquitylation enzymes that are associated with the pathophysiology of Alzheimer’s disease in order to cure patients with neurodegenerative diseases in the future. In my free time, I play with my children, go cycling, and read comics.
I am a PhD student in the Department of Neurosciences and I joined the Wernig lab in the summer of 2020. I hope to study the fundamental cell biology of microglia giving rise to stable tiling in the brain. Furthermore, I am interested in how these mechanisms contribute to brain homeostasis and change with neuronal disease.
During my PhD in Yongjuan Zhao and Honcheung Lee’s lab, I focused on studying the enzymatic activity and function of ADP-ribosyl cyclase, particularly the activity of SARM1 and its role in axon degeneration. This work sparked my interest in neuroscience and neuropathology. Microglia, the resident immune cells in the brain, play critical roles in neuronal function and can be either beneficial or detrimental in the context of neurodegenerative diseases.In Wernig’s lab, I am interested in investigating the function of microglia in aging and Alzheimer’s disease. My current research focuses on understanding the effects of clonal hematopoietic mutations on microglia and their role in the development of Alzheimer’s disease.
I'm excited about diving into the mysteries of cell fate changes at both the cellular and molecular biology levels, using both experimental and computational tools. In the Wernig lab, I'm on a fascinating journey exploring methods to reprogramm cells into specific neuronal fates. Plus, I'm eager to unravel the basic transcriptional regulation codes in these processes.
Alumni




.gif)

Alan Napole was an LSRP working on elucidating the mechanisms of transgene silencing following iN differentiation from pluripotent stem cells. Concurrently, Alan is also working on understanding the role of microglia in multiple sclerosis via EAE mice models. Previously, he was a CIRM scholar working closely with microglia replacement strategies to develop a human mouse microglia chimera for investigation of neuroimmunological diseases. Alan is currently applying to medical school with interests in neuroscience and surgery. Outside of the lab, he enjoys attending music festivals, playing soccer, and cooking. He moved on to grad school in his home town LA. We wish him all the best for his future endeavors!
Angel earned his B.S. in Biology at California State Polytechnic University, Pomona. As an undergraduate he studied the effects of nicotine on diabetic individuals. As a CIRM fellow at Stanford University, he is interested in using microglia cell replacement and stem cell technology as a potential therapeutic for neurological disorders. Unfortunately, COVID19 cut some of the last of his time he was planning to stay with us. We are proud of Angel that he got into the PhD program at UC Irvine where he moved during these challenging times of natural disasters of the year 2020. Good luck Angel!
Bahareh's research interests were focused on epigenetic regulation of cognition at the level of synapses, using iN system. In my free time she likes to do sports and outdoor activities, cook healthy food, make mixed drinks, play music, and collect vinyl records. After graduating from our Stem Cell and Regenerative Medicine PhD program she moved on as postdoctoral fellow to Boston.
Cheen Euong Ang was a bioengineering PhD student in the Wernig lab. He spent his undergraduate career at McGill University, graduating with a B.Sc. in Chemistry with a focus in biological chemistry. Since coming to Stanford, Cheen Euong has been working on investigating the mechanisms of iN reprogramming and applying the iN platform in disease modeling and aging. Other than doing research in the lab, Cheen Euong has participated several scientific outreach programs such as the Stanford Biomedical Engineering Society undergraduate academic mentoring program and Stanford Institute of Medical Summer High School Student Research Program. Cheen moved on to do his postdoc in Xiaowei Zhuang at Harvard University in Cambridge, MA.
Christian's project focused on the function of synaptic proteins in the context of human neurons. Brain research has made tremendous advances in the last decades using animal models. However, most neuropathological conditions, such as autism or schizophrenia, are unique to human physiology. Thus, I study the effect of genetic alterations of synaptic proteins in induced human neurons using immunohistochemistry, analysis of gene and protein expression, as well as electrophysiology. Christian moved on to a senior lecturer position at the newly formed university of Linz, Austria. Good luck there!
Graduating from UC Berkeley majoring in Biology with an honors in Neuroscience, and having worked as an MD at the Royal Melbourne Hospital, I have developed a particular interest in translational neuroscience. In the lab, I am investigating the efficacy of cell therapies for neurological disorders. I am currently working on microglia based therapies for major neurological diseases, including multiple sclerosis.
I am a molecular biologist with a focus on neuro- and cancer biology. I am interested in using human iPSCs to develop new in vitro disease models, particularly for neuropsychiatric disorders and neurooncology. This led me to the Wernig lab where I started my postdoc to explore the advantages of direct neuronal reprogramming and the intricacies of step-wise differentiation of neural cell types. I am genetically engineering iPSC lines to generate complex genomic alterations and inducible mutations. Following differentiation into the corresponding disease-relevant tissue, I am puzzling together cellular phenotypes, transcriptional regulation, protein interactions, and epigenetic changes for a better understanding of the disease biology. In my spare time, I enjoy a new definition of chaos by my 2-year-old daughter.
Hiroko received Ph.D. from UCLA Neuroscience program in Jim Waschek's lab. She's currently a postdoc studying the disease mechanism of pediatric brain disorder Pelizaeus-Merzbacher Disorder, affecting oligodendrocyte development and myelination. She uses patient-derived iPS cells, gene targeting in iPS cells, and animal models.
I was an administrative associate for the Wernig Lab for a brief period in 2019. I am a native San Franciscan and I love the bay area. I received my undergrad from San Francisco State. I am fluent inSpanish so if you ever need a Spanish lesson come by desk. My husband and I have three beautiful childrenand during my time off I really enjoying spending time with my family.
I grew up in Long Island, NY. I attended college at Cornell University majoring in Biological Sciences with a concentration in Neurobiology and Behavior. After graduation in 2009, I moved to Nashville, TN to join the Medical Scientist Training Program at Vanderbilt University where I earned a combined MD/PhD. In 2016, I started my residency in Neurosurgery at Stanford. In the Wernig lab, I am interested in developing microglia-based regenerative therapies for neurodevelopmental and neurodegenerative disorders.
I received my PhD from National Institute of Biological Sciences (NIBS) at Beijing, China. My PhD mentor is Dr. Xiaodong Wang, who is a chinese-born American biochemist best known for his work with cytochrome C on apoptosis. He is a member of United States National Academy of Sciences and Howard Hughes Medical Institute.
During my PhD stage, my major work is to try and find function of a protein named RIP3 in necrosis (other program cell death) pathway. I use human fetal neural stem cells to study mutation caused epigenetic rigidity in reprogramming process. A lot of patient diseases are caused by gene mutation, but we do not know which kind of cells and which site of mutate will cause disease. We try to work on neural stem cells to induce some point mutation to imitate brain cancer disease from reported mutation site in human patient. Then we will know which mutation site is the key and try to fix it by the biology method.
The immense potential of genetic engineering technology as a tool to understand disease mechanisms and as a therapy motivates me to pursue research. I am studying CRISPR/Cas9 mediated gene correction of mutations in patients suffering from Epidermolysis bullosa. I am also studying iPSC-derived induced neurons to model neurological disorders.
Minjeong was the administrative associate for the Wernig Lab. We all loved her positive spirit that made our lab a happy place. We were fortunate to have her for a full year! During her time off, she loved hanging out with my family and trying out different recipes. She moved on to a full time position in business.
Dr. Moritz Mall is a postdoctoral fellow in the lab. After studying biochemistry and molecular biology at the LMU in Munich and the ETH in Zurich he received his Ph.D. from the EMBL in Heidelberg for his mechanistic studies on mitotic cell division. Besides surfing Moritz’s passion is to understand the molecular mechanisms of cell fate determination during reprogramming, development and disease.
A striking feature of the mammalian nervous system is its enormous cellular diversity. The biological question that drives my research in long-term is to understand how the nervous system develops and achieves its extraordinary complexity. I would also like to leverage my expertise in lineage reprogramming, stem cell biology, and neurobiology to develop reprogramming-based human cell culture models and study the fundamental processes underlying the development and function of human nervous system under normal and pathological conditions. Outside the lab, I like spending my time cycling, rock climbing and hiking.
I grew up in Australia where I completed an undergraduate degree in neuroscience and an MD at the University of Melbourne, Australia. I then completed my intern year at the Royal Melbourne Hospital. I have a keen interest in stem cell therapeutics and their potential application in treating debilitating neurological conditions. My current research focus is on novel microglia based therapies that can potentially ameliorate disease progression in multiple sclerosis.
With billions of neurons interconnected with many billions of synapses, our brain is the most complex object in the known universe. I like to think that I’m doing my part in understanding it. I use human neurons to study a synaptic protein called Neuroligin 4 that is not found in mice, therefore very complicated to study but at the same time extremely fascinating. When mutated Neuroligin 4 causes Autism and impairs the synaptic transmission of neurons. After leaving his prominent imprint on the lab he moved on to join the faculty at Mount Sanai School of Medicine in New York. Good luck Samuele, we will miss you!
My research interest is to investigate how genetic background contributes to the pathomechanism of complex mental disorders such autism or schizophrenia. In order to achieve my goals I am using various experimental techniques including rodent and human neuronal cell culture preparations, immunohistochemistry, gene and protein expression analysis and electrophysiology. As a scientist, my motto is: "We work in the dark to serve the light." Tamas moved back to his home country Hungary.
Virginia is a dentist that worked with underserved communities before taking time off to care for her family. She is happy to be the administrative associate for the Wernig lab. In her free time she enjoys reading, playing tennis, and spending time with her family. Unfortunately, due to COVID19 Virginia needed more time to care for her children and had to leave us. She was a delight to have around and we will dearly miss her!
Wendy received her Ph.D. from Columbia University, where she studied how cholesterol-rich membrane microdomains regulate a distinct function of a transmembrane protein, resulting in specific behavioral outcomes. In the Wernig lab, she aimed to understand the molecular mechanisms by which neuronal identity is established and how its disruption leads to neurological diseases focusing on the gene Myt1l.
Yingfei Liu is a Neurobiology PhD candidate from Xi`an Jiaotong University,China. She is studying here for two years as a Joint-Training Doctoral Program. Her previous research interest was the self-renewal and differentiation of the neural stem cells. Now She is studying the mechanisms of direct lineage reprogramming of fibroblasts into neurons. Yingfei was great to have in the lab. COVID19 also affected Yingfei's carreer. In the midst of great progress COVID19 precluded much of her continuing to work. She went back to China to arrange for her PhD defense and we are hoping to be able to continue to work with her in the future. Good luck Yingfei!
I completed my PhD in Dr. TY Chang’s lab at Dartmouth College, where I studied cholesterol metabolism in health and disease. In the Wernig lab, my research focus is on studying neurological diseases in human neurons and other neural lineages. I am also interested in developing novel therapeutic approaches for treating incurable neurological disorders using reprogramming technology.
Yongjin received his Ph.D. in functional genomics in 2018 from the Seoul National University, South Korea. For his Ph.D., he focused on genetic mutations of human neurological patients and its functional impacts. Yongjin joined the Wernig lab in October 2018. In the Wernig lab, he is interested in developing therapeutic methods for neurological patients and understanding disease mechanisms using human stem cells and neurons. We are proud that Yongjin accepted an offer to join the faculty at the prestigious Seoul University.
Press
Wernig Lab Protocols & Recipes
Contact
We are always interested to hear from ambitious scientists and potential collaborators.
Marius Wernig
wernig@stanford.edu
Amy Lang, Lab Admin
alang2@stanford.edu
Institute for Stem Cell Biology and Regenerative Medicine
Wernig Laboratory
265 Campus Drive G3141
Stanford, CA 94305
U.S.A.
